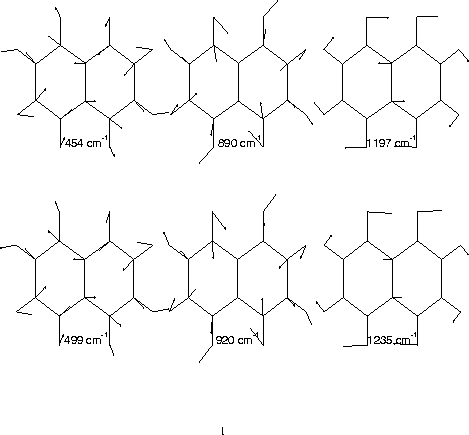
B3g normal modes of naphthalene. Top: S1 CIS/6-31G*. Bottom: S0 HF/6-31G*.
G. Jas and K. Kuczera
Chem. Phys. (1997), 214, 229-241.
Normal mode calculations are presented for the lowest singlet excited states S1 of benzene, naphthalene, anthracene. Optimized geometries and Cartesian harmonic force constants of the excited states are obtained from ab initio calculations at CIS/6-31G, CIS/6-31G*, CIS/6-311G, CIS/6-311G* levels for benzene, and CIS/6-31G and CIS/6-31G* levels for naphthalene and anthracene. Normal mode analysis is performed in internal coordinates, yielding vibrational frequencies and forms of normal modes for the parent molecules and their perdeuterated derivatives. The results are compared with corresponding properties of the ground state S0 calculated at the Hartree-Fock level and with available experimental data. The overall changes in molecular geometry and vibrational spectra upon S0 -> S1 excitation are small. For benzene we find excellent agreement of the entire calculated S1 vibrational spectrum with experimental results. For naphthalene and anthracene the calculated vibrational frequencies are in good agreement with the limited available experimental data. Our calculations provide predictions of frequencies and types of normal modes for the complete S1 vibrational spectra of these molecules, and suggest reassignments of several normal mode frequencies compared to the limited experimental results that are currently available.
B3g normal modes of naphthalene. Top: S1 CIS/6-31G*. Bottom: S0 HF/6-31G*.
_______________________________________________________________________________
Y. Wang and K. Kuczera
J. Phys. Chem. B, (1997), 101, 5205-5213,
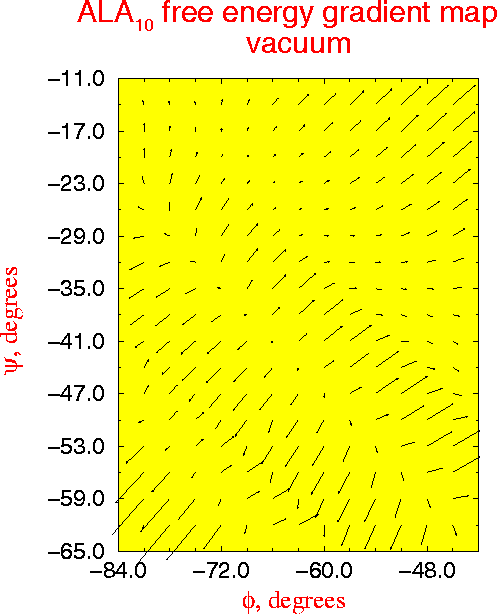
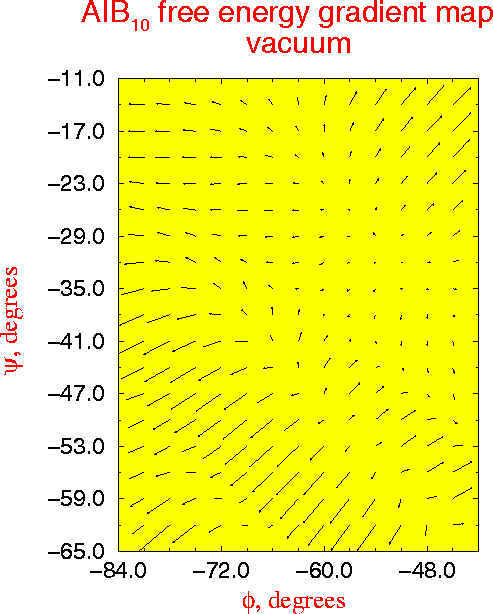
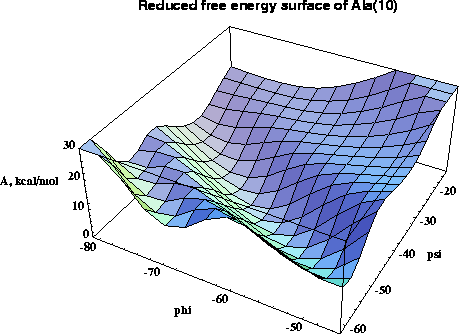
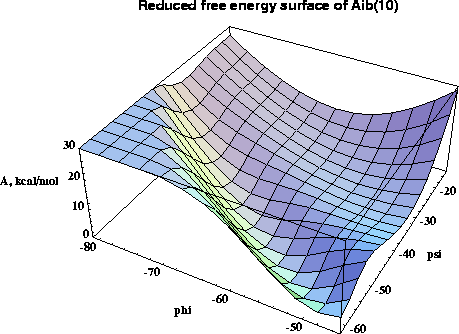
A new multidimensional thermodynamic integration method is applied to investigate free energy surfaces of alanine (Ala) and alpha-methylalanine (Aib) homopeptides in the helical region. In this approach a single molecular dynamics simulation with all phi and psi dihedrals kept fixed yields the free energy gradient with respect to all the fixed conformational coordinates. For regular structures of model peptides (Ala)n and (Aib)n , n=6,8,10 in vacuum, free energy maps in phi-psi space are calculated, and used to roughly locate free energy minima. For (Ala)n alpha-helical minima are found for n=6,8,10 and a pi-helical minimum exists for n=10, while all studied (Aib)n peptides have stable alpha- and 310-helical states. The locations of the free energy minima are further refined by the novel procedure of free energy optimization by steepest descent down the gradient, leading to structures in excellent agreement with experimental data. The stability of the minima with respect to deformations is studied by analysis of second derivatives of the free energy surface. Analysis of free energy components and molecular structures uncovers the molecular mechanism for the propensity of Aib peptides for the 310-helix structure in the interplay between the quality and quantity of hydrogen bonds. The (Ala)n alpha-helix is favored over the 310-helix by all energy terms, exhibiting lower internal strain, lower van der Waals repulsion, and more favorable electrostatic interactions. Although the 310-helix has one more hydrogen bond, in (Ala)n each individual helical hydrogen bond is weaker in the 310-helix than in the alpha-helix. In (Aib)n peptides the added bulk of the alpha-methyls subtly deforms the helices, inducing larger internal strain and larger van der Waals repulsion than in corresponding (Ala)n structures, and making the (Aib)n hydrogen bond geometry better in the 310- than in the alpha-helix . The synergistic effect of greater number of hydrogen bonds and improved interactions within each bond strongly stabilizes the (Aib)n 310-helix making it the favored structure for short peptides.
| 
|
| 
|
_______________________________________________________________________________
G. S. Jas, Y. Wang, S. W. Pauls, C. K. Johnson and K. Kuczera
J. Chem. Phys., (1997), 107, 8800-8812,
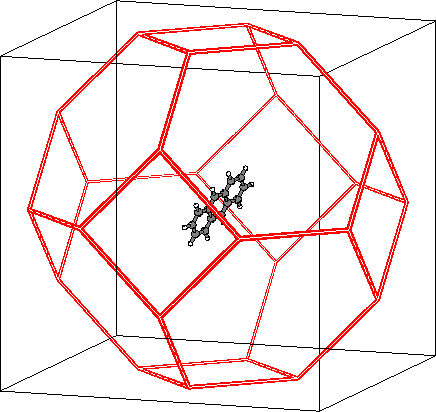
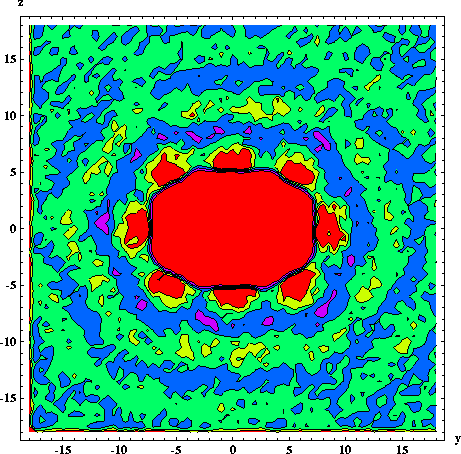
Molecular dynamics simulations and fluorescence anisotropy decay measurements are used to investigate the rotational diffusion of anthracene in two organic solvents - cyclohexane and 2-propanol - at several temperatures. Molecular dynamics simulations of 1 ns length were performed for anthracene in cyclohexane (at 280, 296 and 310 K) and in 2-propanol (at 296 K). The calculated time constants for reorientation of the short in-plane axis were 7-9 and 11-16 ps at 296 K in cyclohexane and 2-propanol, respectively, in excellent agreement with corresponding fluorescence depolarization measurements of 8 and 14 ps. The measured rotational reorientation times and the calculated average rotational diffusion coefficients varied in accord with Debye-Stokes-Einstein theory. Their magnitudes were close to values predicted for an ellipsoid of shape and size equivalent to an anthracene molecule, and exhibited predictable variation with external conditions -- increasing with temperature and decreasing with solvent viscosity. However, analysis of the calculated rotational diffusion coefficients for the individual molecular axes gave a more complex picture. The diffusion was highly anisotropic and changes in temperature and solvent type led to nonuniform variation of the diffusion coefficients. The nature of these changes was rationalized based on analysis of variation of solvation patterns with temperature and solvent.
| 
|
| Anthracene in simulation cell based on cube with side a = 34 A. | Distribution of cyclohexane around anthracene, 296 K, 1 atm. |
_______________________________________________________________________________