
E. Barth, K. Kuczera, B. Leimkuhler and R.D. Skeel
J. Comp. Chem., 16 (1995) 1192-1209.
In molecular dynamics simulations, the fastest components of the potential field impose severe restrictions on the stability and hence the speed of computational methods. One possibility for treating this problem is to replace the fastest components with algebraic length constraints. In this article the resulting systems of mixed differential and algebraic equations are studied. Commonly used discretization schemes for constrained Hamiltonian systems are discussed. The form of the nonlinear equations is examined in detail and used to give convergence results for the traditional nonlinear solution technique SHAKE iteration and for a modification based on successive overrelaxation (SOR). A simple adaptive algorithm for finding the optimal relaxation parameter is presented. Alternative direct methods using sparse matrix techniques are discussed. Numerical results are given for the new techniques, which have been implemented in the molecular modeling software package CHARMM and show as much as twofold improvement over SHAKE iteration.
_______________________________________________________________________________
_______________________________________________________________________________
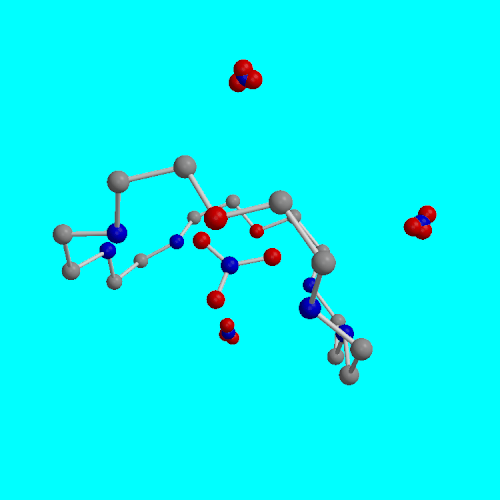
G. Papoyan, K. Gu, J. Wiórkiewicz-Kuczera, K. Kuczera and K. Bowman-James
J. Am. Chem. Soc., (1996), 118, 1354-1364.
Nitrate complexes of two different polyammonium macrocycles, the tetrahydrogen nitrate salts of 1,4,7,16,19,21- hexaaza-10,13-dioxacyclotetracosane [24]N6O2.4HNO3, 1 and 1,4,10,13-tetraaza-7,16-dioxacyclooctadecane [18]N4O2.4HNO3, 2, have been isolated and their structures determined by X-ray crystallographic methods. Compound 1 crystallizes in the orthorombic space group Imma with unit cell dimensions a = 22.692(3) A, b = 19.563(3) A, c = 7.005(1) A, V = 3110(1) A3, and Z = 4. Compound 2 crystallizes in the orthorombic space group Pbca with unit cell dimensions a = 13.877(1) A, b = 15.553(1) A, c = 10.742(1) A, V = 2314.6(5) A3, and Z = 8. Full-matrix least-squares refinement resulted in R = 0.051 and Rw = 0.067 for 1, and R = 0.063 and Rw = 0.072 for 2. Compound 1 has a boat-shaped geometry, and one of the four nitrates is situated in the macrocyclic cavity. Compund 2 is relatively flat with two nitrates above and two below the plane of the molecule. Molecular dynamics simulations were performed for the two molecules using the structural coordinates obtained from the crystal structures and the CHARMM molecular model. Simulations were carried out to 400 ps for 1 and 200 ps for 2 using 2 fs timesteps. The average temperature during the simulations was 302 +- 5 K and the average total energy was -7805.9 +- 0.5 kcal/mol for 1 and -7851.9 +- 0.5 kcal/mol for 2. The conformation of 2 did not change appreciably during the simulation, whereas 1 flattened out considerably due to hydration effects.
|
|
|
| [24]N6O2 average structure from 0-120 ps molecular dynamics | [18]N4O2 average structure from 0-20 ps molecular dynamics |
_______________________________________________________________________________
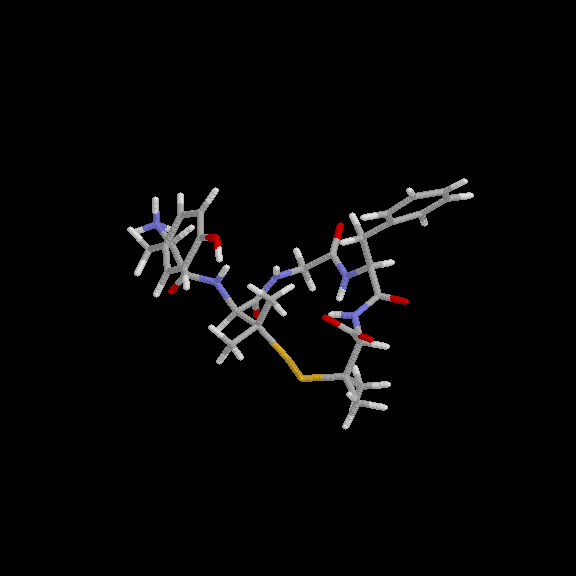
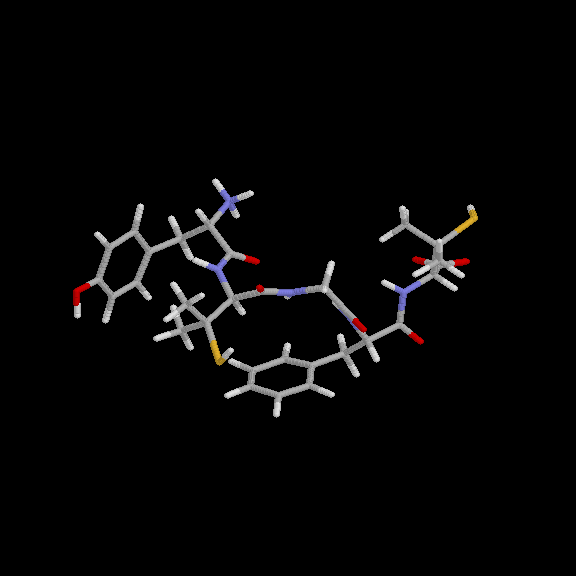
Y. Wang and K. Kuczera
J. Phys. Chem. (1996), 100, 2555-2563,
Three 1 simulations of the solvated DPDPE peptide have been performed, one for the cyclic, and two for the linear form. The trajectories allow us to describe the conformations explored by DPDPE and DPDPE(SH)2 in aqueous solution on the 1 time scale, quantify the conformational constraints imposed by the presence of the disulfide bond by comparing the conformational flexibility of the cyclic and acyclic peptides, evaluate several physical properties: NMR vicinal coupling constants, diffusion coefficients and dipole moments of the peptides, as well as to suggest relations between the calculated properties and biological function. The cyclic peptide, while retaining the general structural features found previously -- presence of a hydrophobic and a hydrophilic face and a parallel arrangement of peptide dipoles, explores four major conformers during the 1 simulation. In two independent simulations, started from an extended and a cyclic-like structure, the linear peptide is found to be about twice as structurally flexible as the cyclic form. Both DPDPE(SH)2 trajectories converge to essentially the same final structure, a type IV beta-turn. This indicates both that a stable, representative conformation of the linear peptide has been found and that the cyclic-like structure is unfavorable when the disulfide bond is not present.
| 
|
| A structure of cyclic DPDPE from 1 ns simulation in water | A structure of linear DPDPE(SH)2 from 1 ns simulation in water |
_______________________________________________________________________________
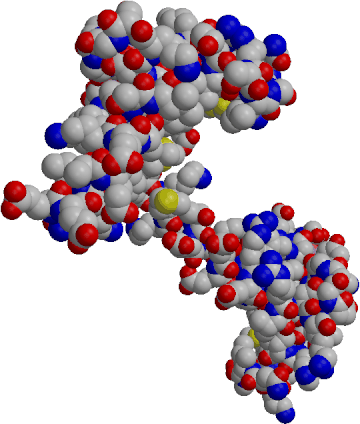
Y. Yao, D. Yin, G. Jas, K. Kuczera, T.D. WIlliams, C. Schöneich
and T.C. Squier
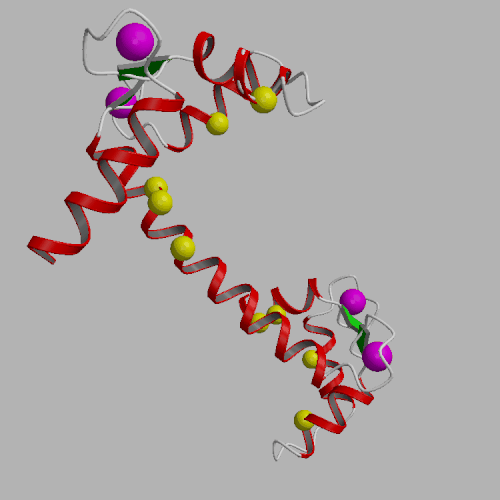
In order to investigate the possibility that calmodulin (CaM) may be a principal target of reactive oxygen species (ROS) produced under conditions of oxidative stress, we have examined wheat germ CaM for the presence of highly reactive sites that correlate with the loss of function. Using reversed-phase HPLC and FAB mass spectrometry after proteolytic digestion, we have identified the sites of modification by hydrogen peroxide. We find that one of the vicinal methionines (i.e. Met146 or Met147) near the C-terminus of CaM is selectively oxidized. The ability of CaM to bind and to activate the plasma-membrane (PM)-Ca-ATPase from erythrocytes was measured. There is a 30-fold decrease in the calcium affinity of oxidatively modified CaM while there is little change in the binding constant between the carboxyl-terminal domain of calcium-saturated CaM and a peptide homologous to the autoinhibitory sequence of the PM-Ca-ATPase, we find that there is a 9-fold reduction in the affinity of the amino-terminal domain of CaM with respect to the ability to bind target peptides. The extent of oxidative modification to one of the vicinal methionines near the carboxy-terminal domain correlates with the loss od CaM-dependent activation of the PM-Ca-ATPase. The presence of the oxidatively modified CaM prevents native CaM from activating the PM-Ca-ATPase, indicating that the oxidatively modified CaM binds to the autoinhibitory sequence of the Ca-ATPase in an altered non-productive conformation. We suggest that the functional sensitivity of CaM to oxidation of one of the vicinal methionines permits CaM to serve a regulatory role in modulating celular metabolism under conditions of oxidative stress. The predominant oxidation of a methionine near the carboxyl terminal of CaM is rationalized in terms of the enhanced solvent accessibility of these vicinal methionines.
| 
|
| Calmodulin CPK representation, PDB structure 3CLN | Calmodulin secondary structure, Ca2+ ions shown as purple spheres, Met CA atoms as yellow spheres (3CLN) |
_______________________________________________________________________________
K. Kuczera
Biopolymers (1996), 39, 221-242,
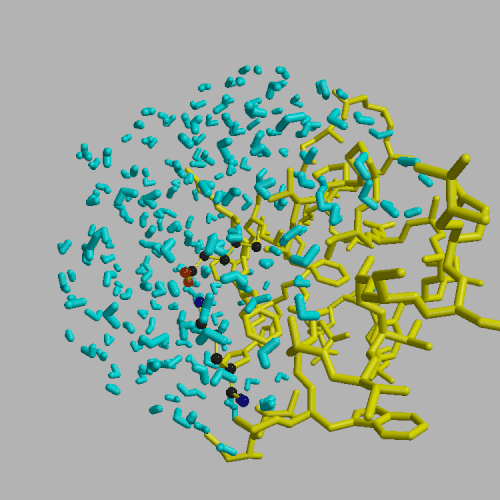
Molecular dynamics simulations have been used to investigate the thermodynamic stability of axial contacts in sickle-cell hemoglobin (HbS). Free energy changes were evaluated for the point mutation beta 121 Glu -> Gln in the axial contact region of HbS crystals. The calculations predict a free energy change of -3.6 kcal/mol per contact for the mutation, which is in qualitative agreement with experimental observations of aggravated sickling found in the double mutant Hb D Los Angeles (beta 6 Glu -> Val, beta 121 Glu -> Gln) relative to HbS beta 6 Glu -> Val). The beta 121 Glu is sequestered in a salt link with beta 17 Lys located on the same polypeptide chain, making the Glu interactions with its surroundings similar in aggregates and individual hemoglobins. Due to this cancellation of the large electrostatic Glu contributions, the weak non--specific interactions between the Gln and the neighboring polypeptide chain are the main contributing factor to the enhanced aggregation of Hb D Los Angeles relative to HbS. Together with the previous study of the lateral contact ( K. Kuczera et al., Proc. Natl. Acad. Sci. USA (1990), 87:8481--8485), the present results provide a more complete picture of the forces driving the sickling aggregation. A comparison of different treatments of internal flexibility in free energy simulations and analysis of rate of convergence of the different calculated properties has also been performed.
 |
| Figure shows four deoxy-HbS molecules; top two and bottom two arranged in parallel strands. Lateral contacts involving beta 6 Val occur at interface between the two strands (red and yellow spheres). Axial contacts involving beta Glu 121 occur within each strand (purple and cyan spheres). Beta chains of strand #1 : yellow, strand #2 : red. |
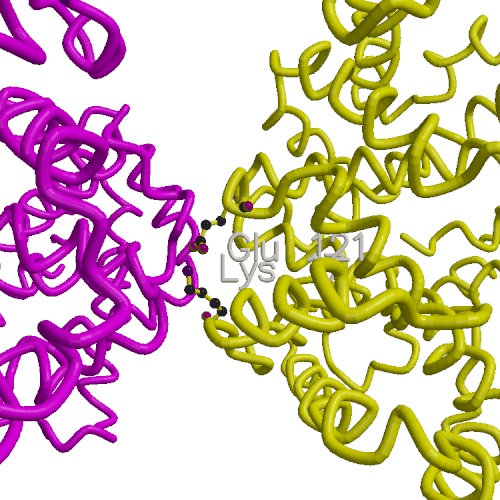
| 
|
| Schematic view of axial contact in strand #2. | System for free energy simulation of uncomplexed axial contact site. |
_______________________________________________________________________________
K. Kuczera
J. Comp. Chem. (1996), 17, 1726-1742.
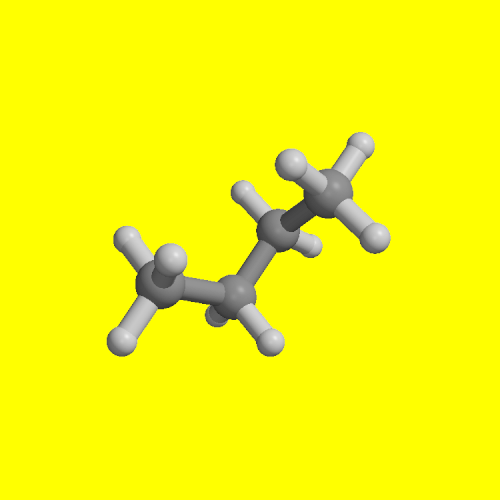
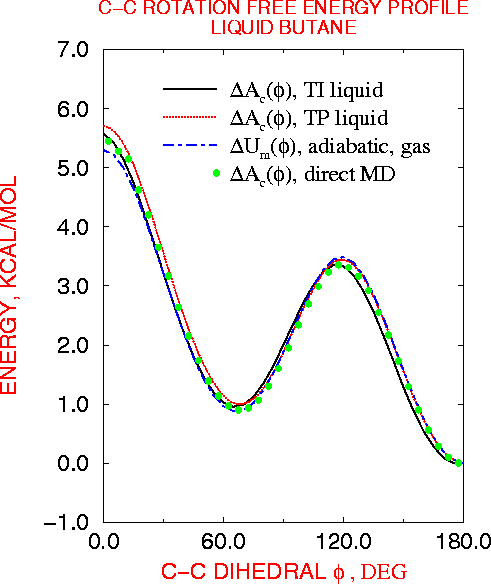
The formal properties of the thermodynamic integration approach to computer simulations of molecular conformational free energy are presented. It is shown how derivatives of free energy with respect arbitrary internal coordinates (e.g. distances, angles, dihedrals) may be evaluated directly as the averages of the corresponding derivatives of the potential energy function and how integration of the derivatives yields free energy profiles, i.e. the dependence of free energy of the system on the chosen coordinates. Schemes are also developed for decomposing the total free energy into energetic and entropic components, into contributions from different potential energy terms - internal, van der Waals and electrostatic, and from different parts of the system - solvent, surrounding chemical groups. This component analysis is the main advantage of the proposed method over existing approaches, providing a novel, powerful tool to improve our basic understanding of conformational equilibria of flexible molecules in condensed media. The method is applied to calculate the free energy profile for rotation around the central C--C bond of butane in the gas phase and aqueous solution. The feasibility of multidimensional calculations is also examined by calculating free energy gradients with respect to all phi and psi dihedrals of decaalanine and deca-alphamethylalanine.

| 
| 
| 
|
| cis transition state | gauche conformer | transition state | trans conformer |
| phi = 0 | phi = 70 | phi = 120 | phi = 180 |
_______________________________________________________________________________
K. Kuczera
in ``Recent Developments in Theoretical Studies of Proteins'', edited by Ron Elber. World Scientific, Singapore, 1996, pp. 1-63. Series: ``Advances in Physical Chemistry'', vol. 7.
An overview of computer simulations of globins -- mostly myoglobin and hemoglobin -- is presented. These results describe the current understanding of the fundamental physico--chemical properties of globins at the microscopic level, including equilibrium fluctuations, nonequilibrium relaxation, and thermodynamics of association and quaternary structural change. The review contains an introduction to modern methods of macromolecular simulation, their application to globins, as well as a discussion of the insights from simulations into biological function.

| 
|
| six-liganded heme from PDB file 1MBC | five-liganded heme from PDB file 1MBD |
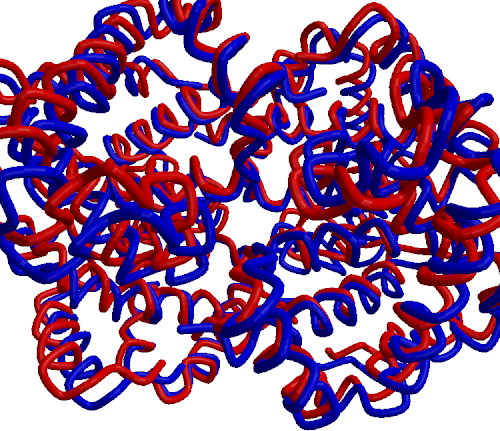
|
| Comparison of oxyhemoglobin (red) and deoxyhemoglobin (blue) structures |
_______________________________________________________________________________
Go back to main abstracts page