|
|
| Sample structures of angiotensinn I (A) and II (B). | Clusters from angiotensin I MD |
_______________________________________________________________________________
K. Kuczera and P. Kursula.
J. Biomol. Struct. Dyn., 30:45-61 (2012).
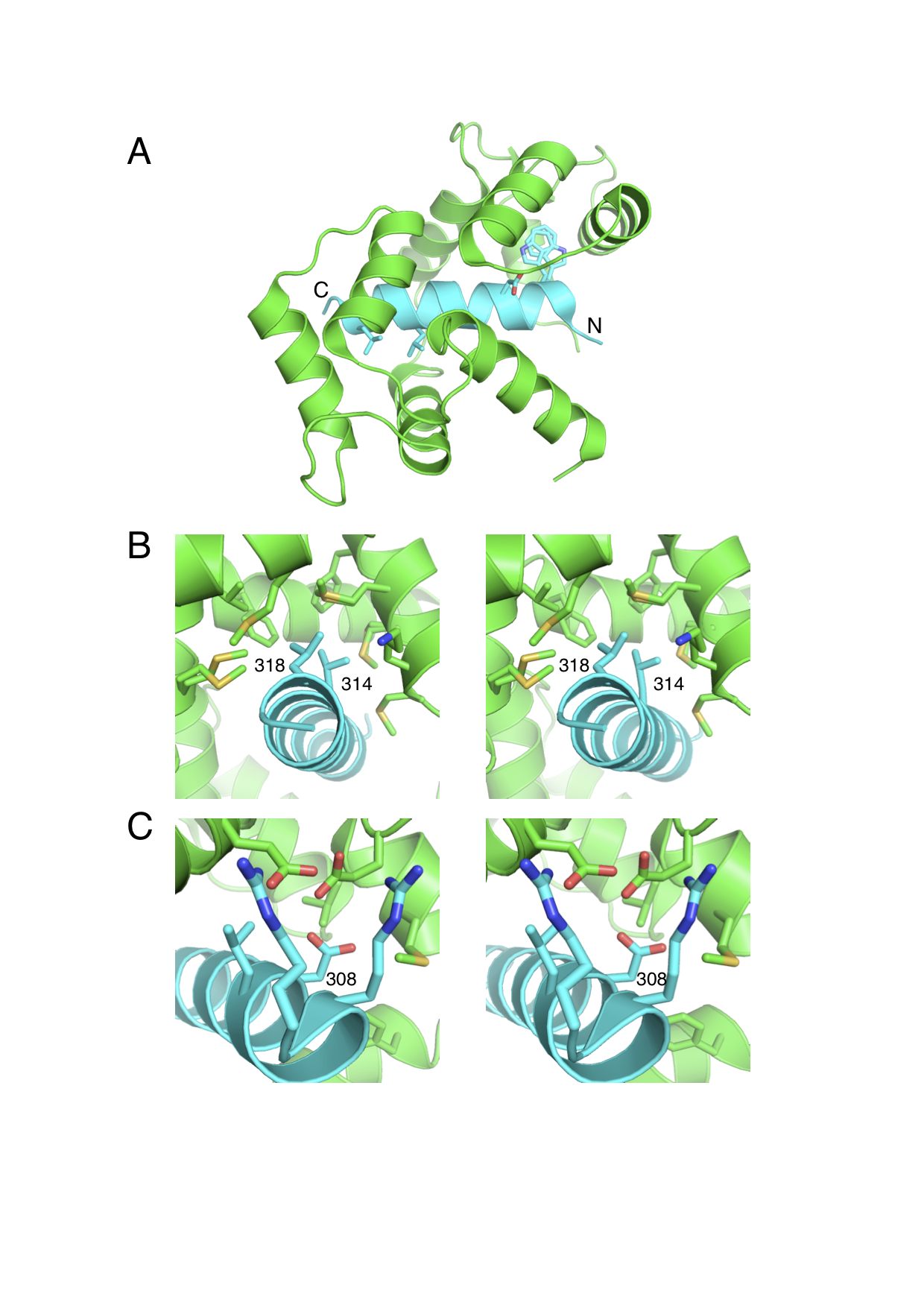
We have studied the interactions between calmodulin and three target peptides from the death-associated protein kinase (DAPK) protein family using both experimental and modeling methods, aimed at determining the details of the underlying biological regulation mechanisms. Experimentally, calorimetric binding free energies were determined for the complexes of CaM with peptides representing the DAPK2 wild type and S308D mutant, as well as DAPK1. The observed affinity of CaM was very similar for all three studied peptides. The DAPK2 and DAPK1 peptides differ significantly in sequence and total charge, while the DAPK2 S308D mutant is designed to model the effects of DAPK2 Ser308 phosphorylation. The crystal structure of the CaM-DAPK2 S308D mutant peptide is also reported. The structures of CaM-DAPK peptide complexes present a mode of CaM-kinase interaction, in which bulky hydrophobic residues at positions 10 and 14 are both bound to the same hydrophobic cleft. To explain the microscopic effects underlying these interactions, we performed free energy calculations based on the approximate MM-PBSA approach. For these highly charged systems, standard MM-PBSA calculations did not yield satisfactory results. We proposed a rational modification of the approach which led to reasonable predictions of binding free energies. All three complexes are strongly stabilized by two effects: electrostatic interactions and buried surface area. The strong favorable interactions are to a large part compensated by unfavorable entropic terms, in which vibrational entropy is the largest contributor. The electrostatic component of the binding free energy followed the trend of the overall peptide charge, with strongest interactions for DAPK1 and weakest for the DAPK2 mutant. The electrostatics was dominated by interactions of the positively charged residues of the peptide with the negatively charged residues of CaM. The nonpolar binding free energy was comparable for all three peptides, the largest contribution coming from the Trp305. About 2/3 of the buried surface area corresponds to nonpolar residues, showing that hydrophobic interactions play an important role in these CaM:peptide complexes. The simulation results agree with the experimental data in predicting a small effect of the S308D mutation on calmodulin interactions with DAPK2, suggesting that this mutation is not a good model for the S308 phosphorylation.
|
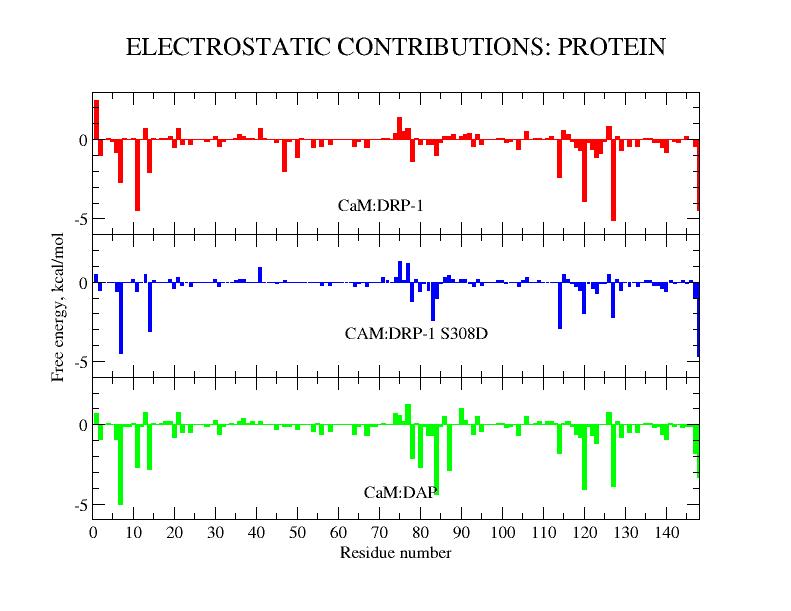
| Crystal structure of CaM:DAPK2(S308D) complex. | CAM residue condtributions to electrostatic binding free energy for CAM:DAPK peptide complexes. |
_______________________________________________________________________________
G.S. Jas and K. Kuczera.
Molecular Simulation, 38:682-694 (2012). Invited article for special issue New Developments in Molecular Simulation.
We present results of microsecond-length simulations of folding for three model helix-forming peptides, penta-alanine Ac-A5-NH2 (acA5a), a modified pentapeptide Ac-WA3H+-NH2 (WH5) and a 21-residue peptide Ac-WA3H+-(AAARA)3-A-NH2 (WH21), employing three protein force fields, AMBER03, CHARMM27 and OPLS-AA. The calculated helix populations are in good agreement with observations for all peptides, and the measured kinetic rate constants of WH5 are well reproduced. Our results yield new and interesting insights into the mechanism of alpha-helix folding. For acA5a, representig a flexible generic helical nucleus, we find significant variation of the folding pathway with employed force field. For the WH5 helix, which includes a stabilizing sidechain interaction, the nucleation step also varies with force field, but an intermediate with first two hydrogen bonds formed is a common feature in all models. For WH21 the three employed force fields also differed in details of the helix initiation stage. However, the main stages of folding, involving first the central region, then the N-terminal and finally the C-terminal, were very similar in AMBER03, CHARMM27 and OPLS-AA. Single-helix structures were dominant intermediates, with a minor contribution from helix-turn-helix conformers. The agreement between simulations and experimental data indicate that current protein force fields provide a reliable description alpha-helix folding.

|

| Structures of simulated peptides. | Unfolding pathway for WH21 from AMBER03 REMD. |
_______________________________________________________________________________
G.S. Jas, W. Hegefeld, P. Majek, K. Kuczera and R. Elber.
J. Phys. Chem. B 116:6598-6610 (2012).
We consider the kinetics and thermodynamics of a helical turn formation in the peptide WAAAH. NMR measurements indicate that the peptide has significant tendency to form a structure of a helical turn, while temperature dependent CD establishes the helix fraction at different temperatures. Molecular Dynamics and Milestoning simulations agree with experimental observables and suggests an atomically detailed picture for the turn formation. Using a network representation two alternative mechanisms of folding are identified: (i) a direct co-operative mechanism from the unfolded to the folded state without intermediate formation of hydrogen bonds and (ii) an indirect mechanism with structural intermediates with two residues in a helical conformation. This picture is consistent with kinetic measurements that reveal two experimental time scales of sub nanosecond and several nanoseconds.
|
|
| Sample anchor structures for WH5 Milestoning. | Milestoning folding pathway for WH5. |