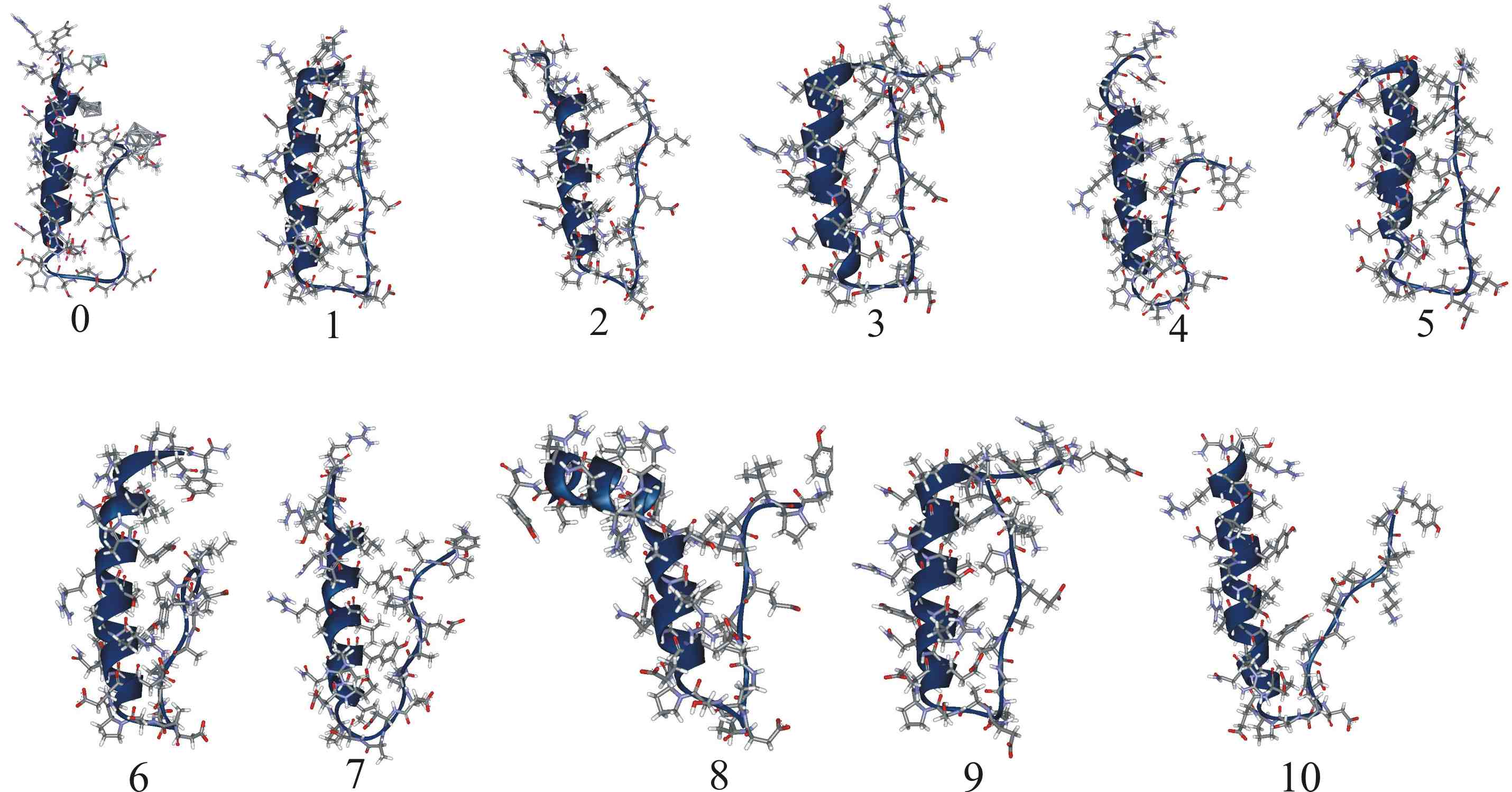
|
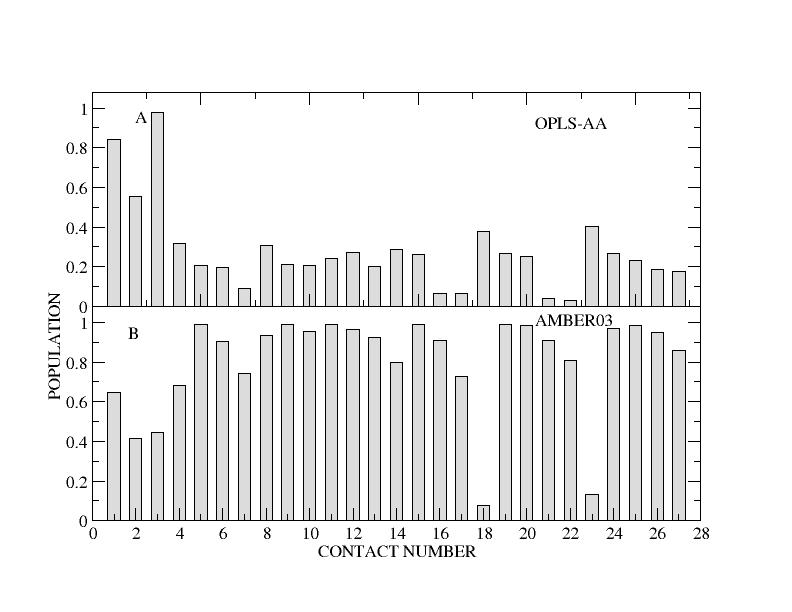
| Sampled structures for hPYY. | Populations of experimentally observed NOE contacts found in hPYY MD. |
_______________________________________________________________________________
S. Parthasarathy, A. Altuve, S. Terzyan, X. Zhang, K. Kuczera, M. Rivera and D.R. Benson.
Biochemistry, 50:5544-5554 (2011).
Mammalian type B (mitochondrial) cytochromes b5 exhibit greater amino acid sequence diversity than their type A (microsomal) counterparts, as exemplified by the type B proteins from human (hCYB5B) and rat (rCYB5B). The comparison of X-ray crystal structures of hCYB5B and rCYB5B reported herein reveals a striking difference in packing involving the five-stranded beta-sheet, attributable to fully buried residue 21 in strand beta4. The greater bulk of Leu21 in hCYB5B in comparison to Thr21 in rCYB5B results in a substantial displacement of the first two residues in beta5, and consequent loss of two of the three hydrogen bonds between beta5 and beta4. Hydrogen-bonding between the residues is instead mediated by two well-ordered, fully buried water molecules. In a 10 ns molecular dynamics simulation, one of the buried water molecules in the hCYB5B structure exchanged readily with solvent via intermediates having three water molecules sandwiched between beta4 and beta5. When the buried water molecules were removed prior to a second 10 ns simulation, beta4 and beta5 formed persistent hydrogen bonds identical to those in rCYB5B, but the Leu21 side chain was forced to adopt a rarely observed conformation. Despite the apparently greater ease of water access to the interior of hCYB5B than of rCYB5B suggested by these observations, the two proteins exhibit virtually identical stability, dynamic and redox properties. The results provide new insight into the factors stabilizing the cytochrome b5 fold.
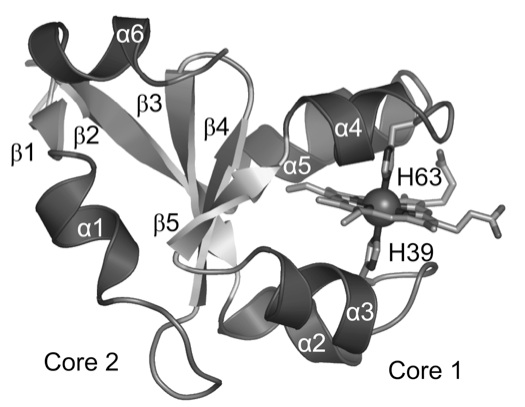
|
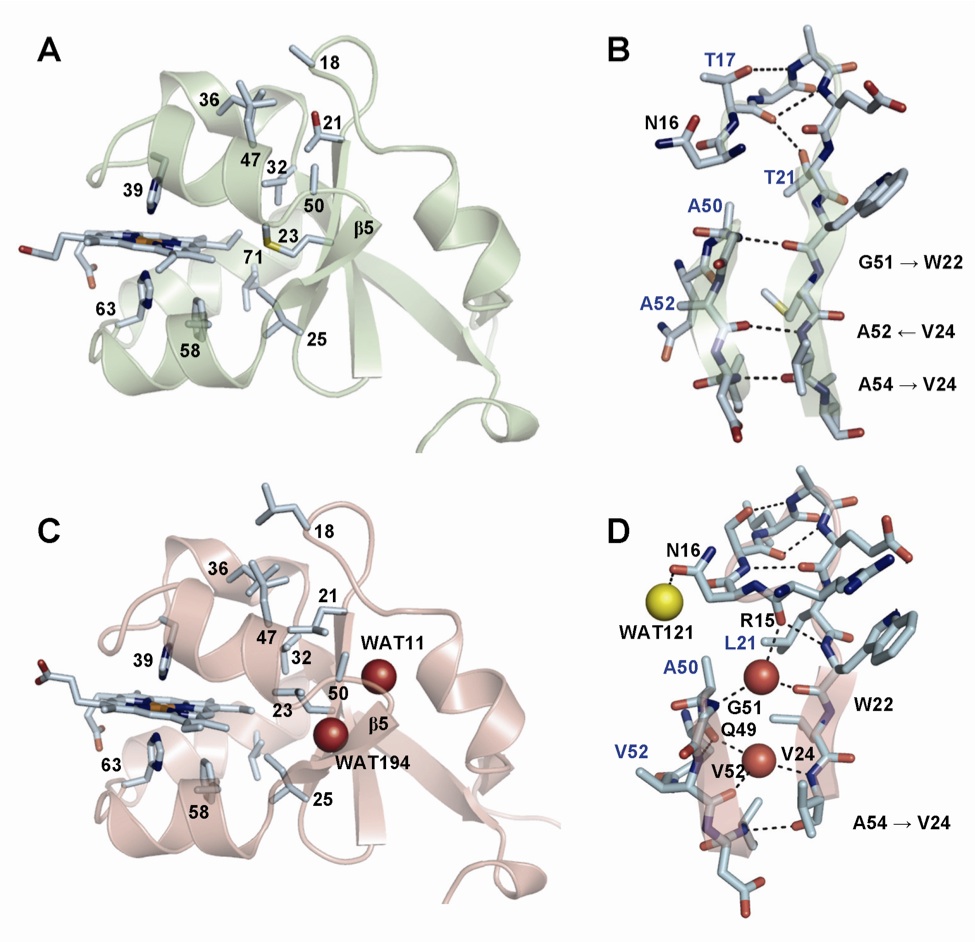
| Schematic structure of cb5. | Comparison of crystal structures of rat (top) and human (bottom) cb5. |
_______________________________________________________________________________
K.-H. Lee, K. Kuczera and M.M. Banaszak Holl.
Biopolymers, 95:182-193 (2011).
Molecular dynamics simulations were carried out to calculate free energy differences between the folded and unfolded states of wild type and mutant collagen model peptides. The calculated stability of the collagen models was compared to the severity of Osteogenesis Imperfecta. Free energy differences of Gly->Xaa (Xaa: Ser, Cys, Glu and Asp) mutations between the wild type and the mutants at position 15 of the model peptide were 3.8, 4.2, 5.6 and 8.8 kcal/mol, respectively. The corresponding free energy differences of a second Gly mutation at the same position in different chains were on average 1.3, 1.5, 2.9, and 5.4 kcal/mol, respectively. Free energy simulations were also performed to estimate the relative stability between an oxidized form and a reduced form of the mutants containing two Cys residues, which indicated that the mutant of the collagen-like peptide containing an intramolecular disulfide bond was more stable than the mutant containing one Cys residue but less stable than the wild type. The calculated free energy differences between an oxidized and a reduced form of the mutants containing two Cys residues are 0.8 and 2.6 kcal/mol for the disulfide bonds between chain A and B and between chain A and C, respectively