|
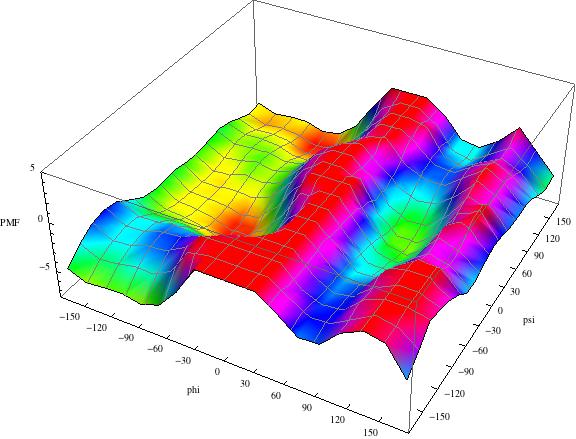
| Blocked Ala5 structure with H-honds. | Phi-psi map of sampled conformations in 1 microsecond MD with OPLS-AA/TIP3. |
_______________________________________________________________________________
K. Kuczera, J. Unruh, C.K. Johnson and G.S. Jas.
J.Phys.Chem. A, 114:133-142 (2010).
We present a study of reorientation dynamics of the three aromatic amino acids and their side chain models in aqueous solution. Experimentally, anisotropy decay measurements with picosecond time resolution were performed for blocked tryptophan, tyrosine and phenyalanine and model compounds p-cresol and 3-methylindole. Computationally, rotational diffusion was modeled by molecular dynamics simulations for the three aromatic residues and their side chain models: benzene, toluene, phenol, p-cresol, indole and 3-methylindole in explicit water. Our simulations used the CHARMM protein force field and associated TIP3P water model and tend to overestimate the rotational correlation times. However, the simulations yield several interesting qualitative insights into reorientational motions that complement the experimental measurements. The effects of substituent and temperature on reorientations of the parent compounds are well reproduced computationally. Additionally, simulations indicate strongly anisotropic reorientations for most of the studied compounds and a separation of time scales between conformational dynamics and rotational diffusion. Comparison with continuum hydrodynamic models suggests that we may consider that the blocked amino acids move under stick boundary conditions, while dynamics for most of the model compounds falls between stick and slip conditions. Our systematic treatment of blocked amino acids, starting from the parent compounds - benzene, phenol and indole, provides a baseline for understanding the anisotropy decay signals of more complicated peptide systems.

|
| Structures of sidechains and models. | Simulated correlation functions for Trp model reorientations. |
_______________________________________________________________________________