_______________________________________________________________________________
Conformational
Heterogeneity of a Leucine Enkephalin Analog in Aqueous Solution and SDS
Micelles: Comparison of Time-Resolved FRET and Molecular Dynamics Simulations.
Unruh, K. Kuczera and C.K. Johnson.
J.Phys.Chem. B, 113:14381-14392 (2009).
We have undertaken time-resolved Foerster resonance energy transfer (FRET) and molecular dynamics simulations to analyze conformations and conformational heterogeneity of an analog of leucine enkephalin in solution and in the presence of SDS micelles. Enkephalins are opioid pentapeptides that interact with opioid receptors in the central nervous system. We used time-correlated single-photon counting to detect energy transfer between the N-terminal tyrosine and a tryptophan residue substituted for phenylalanine at the 4 position. FRET from Tyr to Trp was measured over a temperature range from 5 C to 55 C in aqueous solution. By taking into account Tyr rotamer interconversion rates measured previously, we determined average distances between Tyr and Trp for the two populated rotameric conformations of Tyr. Molecular dynamics simulations (100 ns) support this analysis and indicate extensive conformational heterogeneity. The simulations also predict that the FRET orientational factor is correlated with the Tyr-Trp separation. The possible effect on FRET calculations is discussed. The errors that would be introduced by not accounting for the correlation between orientation and distance appear to be largely offset in YGGWL by a weighting bias inherent in the R-6 dependence of the energy-transfer rate. The Tyr lifetimes decrease upon titration of the peptides with SDS, indicating formation of compact conformations of the peptide in the micelle environment. This result is consistent with the conjecture that the lipid environment may induce formation of bioactive conformations of the peptide.

|

|
Kinetic scheme for FRET with conformational dynamics.
| Sample MD structures of YGGWL showing Tyr conformation and Tyr...Trp distance.
|
_______________________________________________________________________________
Molecular dynamics simulations with milestoning.
K. Kuczera, G.S. Jas and R. Elber.
J. Phys. Chem. A 113: 7461-7473 (2009).
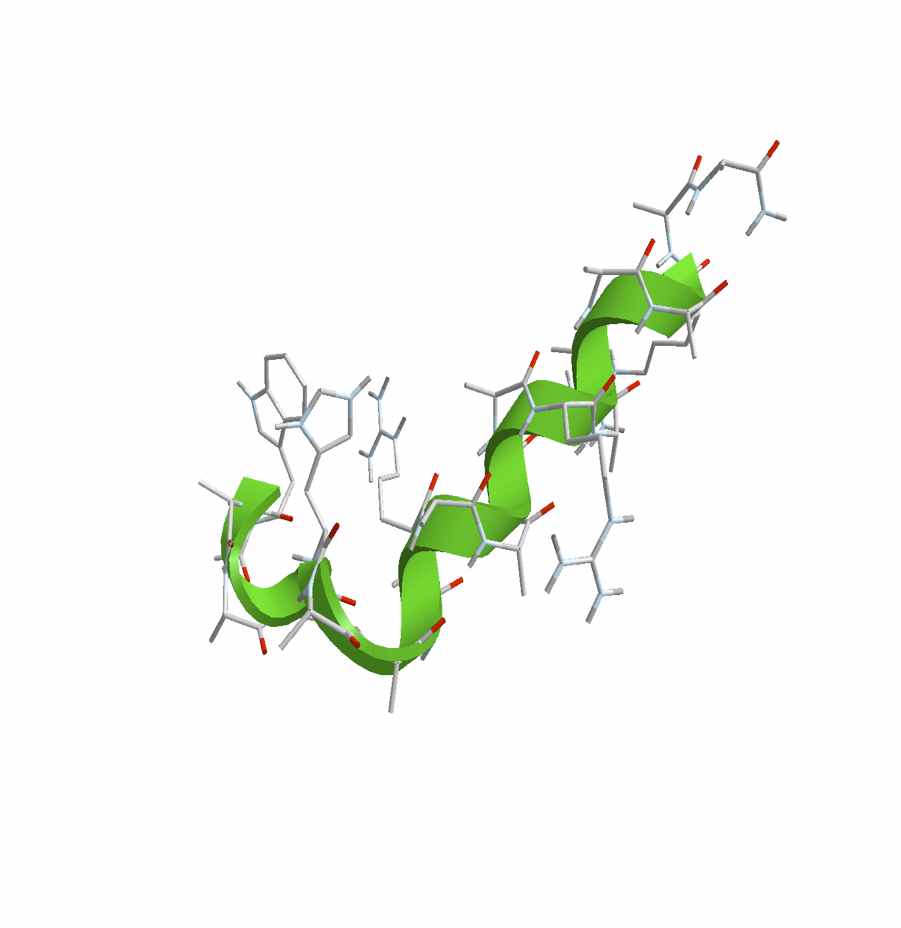
The unfolding process of a helical heteropeptide is studied by computer simulation in explicit solvent. A combination of a functional optimization to determine the reaction coordinate and short time trajectories between “Milestones” is used to study the kinetic of the unfolding. One hundred unfolding trajectories along three different unfolding pathways are computed between all nearby Milestones providing adequate statistics to compute the overall first passage time. The radius of gyration is smaller for intermediate configurations compared to the initial and final states suggesting that the kinetics (but not the thermodynamics) is sensitive to pressure. The transitions are dominated by local bond rotations (the dihedral angle) that are assisted by significant nonmonotonic fluctuations of nearby torsions. The most effective unfolding pathway is via the N-terminal.

|
|
Scheme of setting up Milestones.
| Intermediate structure for unfolding along path 3 (N-terminal unfolding).
|
_______________________________________________________________________________
Go back to main abstracts page
or go on to 2008 abstracts