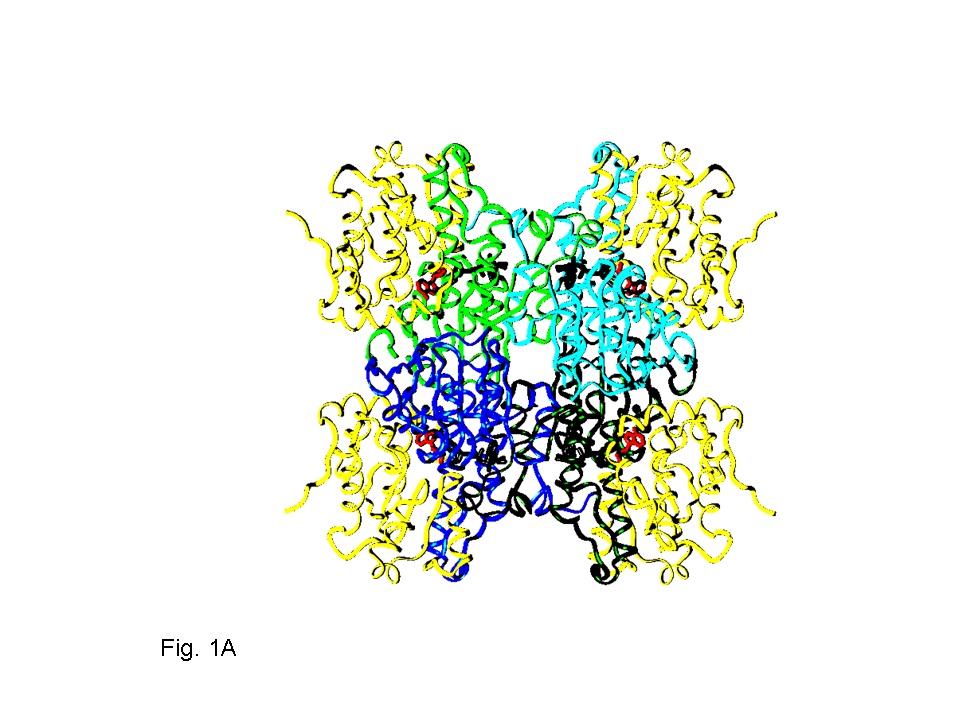
|
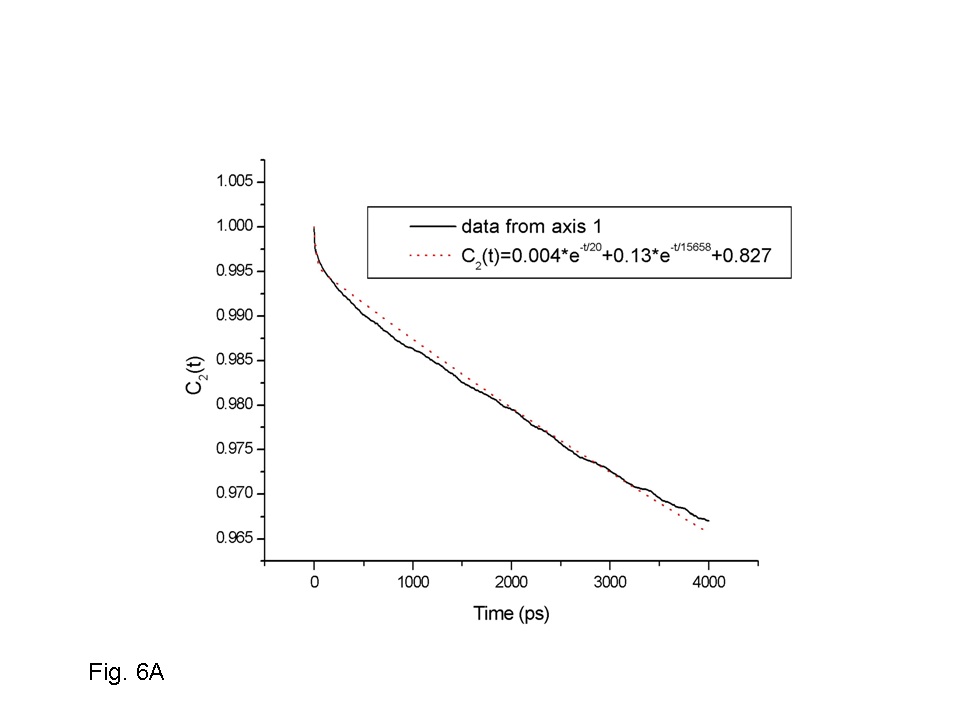
| Structure of SAHH. | Average reorientation of the four SBS from MD. |
_______________________________________________________________________________
Y. Houndonougbo, K. Kuczera and G. S. Jas.
J. Biomol. Struct. Dyn., 26: 17-34 (2008).
We have performed a series of 5-20ns molecular dynamics simulations to investigate the influence of environment and force field on the microscopic structure and dynamics of phospholamban (PLB), a small integral membrane protein. Effect of the environment is studied by simulations in methanol, water and DPPC bilayer, and the influence of force field modification followed by using three variants of the CHARMM22 force field, the standard parameters with nonbonded cutoffs, a version with cutoffs and improved description of protein backbone tortions (CMAP correction), and one with both CMAP correction and Ewald treatment of electrostatics. The simulations show that two main features of PLB structure and dynamics are present under all studied conditions: existence of two well-defined helical domains at the N- and C-termini, and large-amplitude rigid-body motions of these domains. The average interhelix angle of PLB was sensitive to the environment. In the methanol and water solution trajectories, the two helical domains tended to adopt a “closed” orientation, with the interhelix angle below 90 deg, while in the lipid bilayer the domains tend to be in an open conformation, with the interhelix angle above 90 deg. Within each studied environment, simulations employing different force field models provided qualitatively similar description of PLB structure and dynamics. The only significant discrepancy was the presence of -helical hydrogen bonds in trajectories generated with the standard CHARMM22 force field. Trajectories generated with CMAP correction, with both cutoff and Ewald electrostatics, exhibited predominantly -helical and some 310-helical hydrogen bonding interactions, and no -helical hydrogen bonding, in accord with NMR data. Thus, our results indicate that models including CMAP, with both cutoff and Ewald electrostatics, provide the most realistic description of PLB structure and dynamics. Many features observed in the simulations are in a good agreement with the experimental measurements. These include the mostly helical secondary structure of the protein, including the range explored by the interhelix angle in methanol, as well as the interhelix distance and C-terminal helix orientation in the DPPC bilayer. The observed effect of opening up of the PLB interhelix angle in the lipid environment relative to solution is also qualitatively reproduced in the simulations as well as the more rigid and compact structure of the C-terminal domain in the membrane relative to solution.
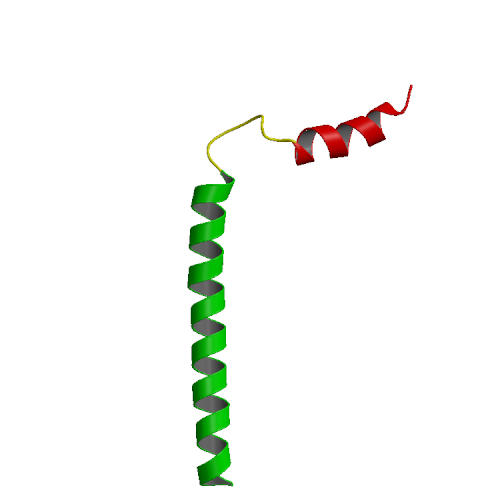
|

| NMR structure of phoshlamban: 1N7L model 1. | PLB in DPPC bilayer with water and Na+/Cl- ions |
_______________________________________________________________________________