M. Wang, R. L. Schowen, R. T. Borchardt and K. Kuczera.
Biochemistry, in press (2005).
The structure and fluctuations of the enzyme S-adenosyl-L-homocysteine hydrolase (SAHH) are analyzed to help explain its biological function. Besides the previously identified open structure, characteristic of the substrate-free enzyme, we find two distinct structures in enzyme-inhibitor complexes, the closed and closed-twisted conformers. Both closed conformers differ from the open form by a hinge bending motion of two large domains within each subunit, which isolate the inhibitor bound in the active site from bulk solvent. The closed-twisted form further differs from the closed form by a rigid body twist of the two-subunit dimers. The local structural fluctuations of SAHH are analyzed by performing block normal mode analysis of the tetrameric enzyme in its three forms. For the open form we find that the four lowest frequency normal modes, corresponding to the collective motions of the protein with the largest amplitudes, are essentially combinations of the hinge bending deformations of the individual subunits. Thus, the mechanical properties of the open structure of SAHH lead to the presence of structural fluctuations in the direction of the open-to-closed conformational transition. A candidate for such a motion has been observed in previous fluorescence depolarization studies of the enzyme. Both structural and normal mode analyses indicate that residues 180-190 and 350-356 form hinge regions, connecting large domains which tend to move as rigid bodies in response to interactions with substrate, intermediates and product of the enzymatic reactions. We propose that these hinge regions play a crucial role in the enzymatic mechanism of SAHH. In contrast to the open form, normal mode calculations for the closed conformations show strong coupling of the hinge bending motions of the individual subunits to each other and to other low-frequency vibrations. Thus, information about structural changes related to reaction progress in one active site may be mechanically transmitted to other subunits of the protein, explaining the cooperativity found in the enzyme kinetics.

|

|
| Animation of open form mode 7 (lowest frequency) | Open form: covariance matrix mode 7 |
_______________________________________________________________________________
M. Wang, J. Zhang, D. Andrei, K. Kuczera, R. T. Borchardt and S. F. Wnuk
Med. Chem. Commun., 48, 3649-3653, (2005).
Moffatt oxidation of 2',3'-O-isopropylidene-L-adenosine and treatment of the resulting crude 5'-aldehyde with hydroxylamine followed by deprotection gave L-adenosine 5'-carboxaldehyde oximes, whose enantiomers are known to be potent inhibitors of S-adenosyl-L-homocysteine hydrolase. The L-adenosine and its 5'-aldehyde oxime derivatives were found to be inactive as inhibitors of AdoHcy hydrolase. Docking calculations using the program AutoDock showed that binding of L-adenosine to AdoHcy hydrolase is both weaker (higher energy) and less specific (larger number of clusters) as compared to D-Ado.
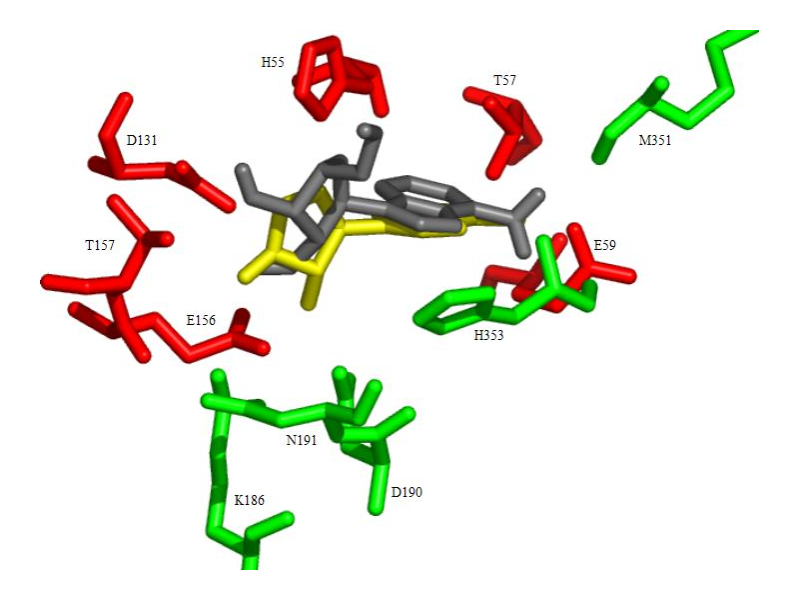
|
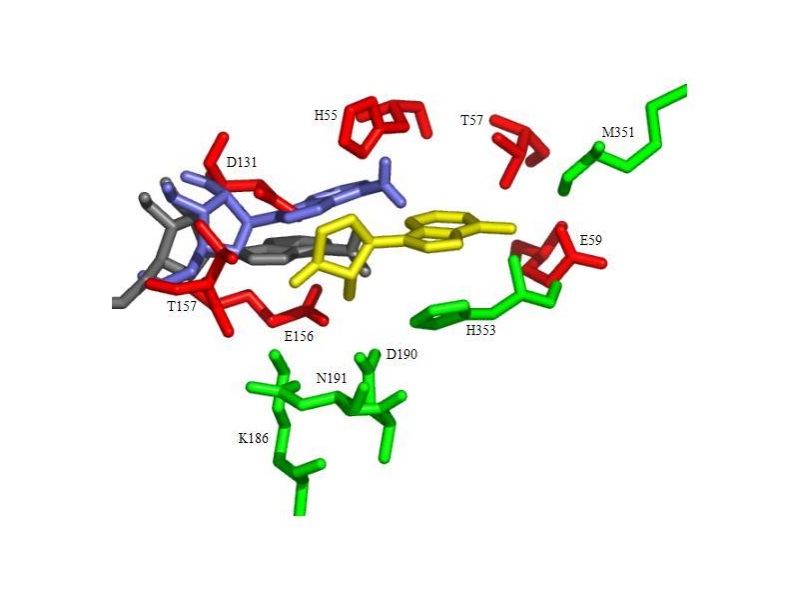
|
| D-Ado docked structures are similar to crystal position of inhibitor DHCeA (gold) | L-Ado docked structures fall outside binding pocket |