|
|
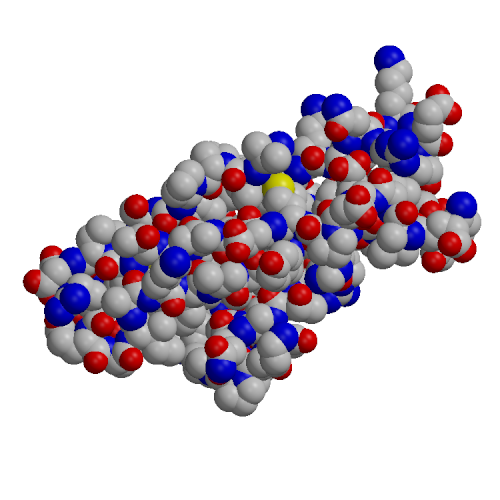
| EC1 domain (1EDH) | EC4 model (based on 1L3W) |
K. Zheng, I. T. Makagiansar, M. Wang, J. L. Urbauer, K. Kuczera and T. J. Siahaan.
Protein Expression and Purification 33, 72-79 (2004).
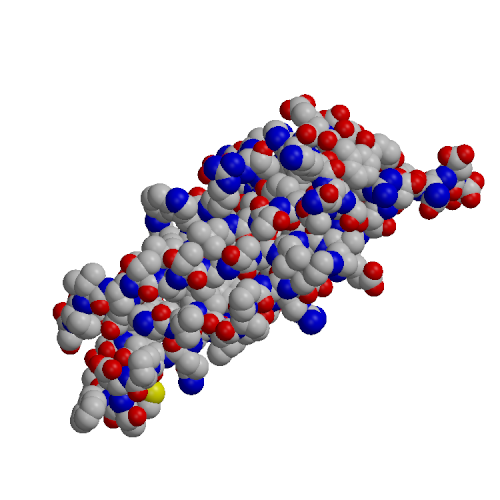
The EC4 domain of E-cadherin (EC4-E) was expressed in E. coli, purified and analyzed by CD and NMR spectroscopy. A homology model of the structure of EC4-E was constructed based on the known X-ray structure of the EC4 domain from C-cadherin (PDB code 1L3W). This model was comapred to the crystal structure of the EC1-E domain (PDB code 1EDH). The EC4 domain tended to higher hydrophilic surface area, lower hydrophobic surface area and fewer hydrogen bonds than EC1. These effects may explain the experimentally observed lower structural stability of the EC4 domain and the decreased tendency of EC4 to form oligomers, relative to EC1.
|
|
|
| EC1 domain (1EDH) | EC4 model (based on 1L3W) |
_______________________________________________________________________________
C. Yang, G. S. Jas and K. Kuczera.
Biochim. Biophys. Acta, 1697, 289-300 (2004).
Calmodulin (CaM) is a small protein involved in calcium signaling; among the targets of CaM are a number of kinases, including myosin light chain kinases (MLCK), various CaM-dependent kinases and phosphorylase kinase. We present results of molecular dynamics simulations of 4 ns length for calmodulin in its three functional forms: calcium-free, calcium-loaded, and in complex with both calcium and a target peptide, a fragment of the smooth muscle MLCK. The simulations included explicit water under realistic conditions of constant temperature and pressure, the presence of counterions and Ewald summation of electrostatic forces. Our simulation results present a more complete description of calmodulin structure, dynamics and interactions in solution than previously available. The results agree with a wide range of experimental data, including x-ray, NMR, fluorescence, cross-linking, mutagenesis and thermodynamics. Additionally, we are able to draw interesting conclusions about microscopic properties related to the protein's biological activity. First, in accord with fluorescence data, we find that calcium-free and calcium loaded calmodulin exhibit significant structural flexibility. Our simulations indicate that these motions may be described as rigid-body translations and rotations of the N- and C-terminal domains occurring on a nanosecond time scale. Our second conclusion deals with the standard model of calmodulin action, which is that calcium binding leads to solvent exposure of hydrophobic patches in the two globular domains, which thus become ready to interact with the target. Surprisingly, the simulation results are inconsistent with the activation model when the standard definitions of the hydrophobic patches are used, based on hydrophobic clefts found in the x-ray structure of calcium-loaded calmodulin. We find that both experimental and simulation results are consistent with the activation model after a re-definition of the hydrophobic patches as those residues which are actually involved in peptide binding in the experimental structure of the calmodulin-peptide complex. The third conclusion is that the calmodulin-peptide interactions in the complex are very strong and are dominated by hydrophobic effects. Using quasi-harmonic entropy calculations, we find that these strong interactions induce a significant conformational strain in the protein and peptide. This destabilizing entropic contribution leads to a moderate overall binding free energy in the complex. Our results provide interesting insights into calmodulin binding to its kinase targets. The flexibility of the protein may explain the fact that CaM is able to bind many different targets. The large loss of conformational entropy upon CaM:peptide binding cancels the entropy gain due to hydrophobic interactions. This explains why the observed entropic contribution to the binding free energy is small and positive, and not large and negative as expected for a complex with such extensive hydrophobic contacts.

|

|

| N-terminal | peptide | C-terminal |
_______________________________________________________________________________
G. S. Jas and K. Kuczera.
Biophysical J., 87 in press (2004).
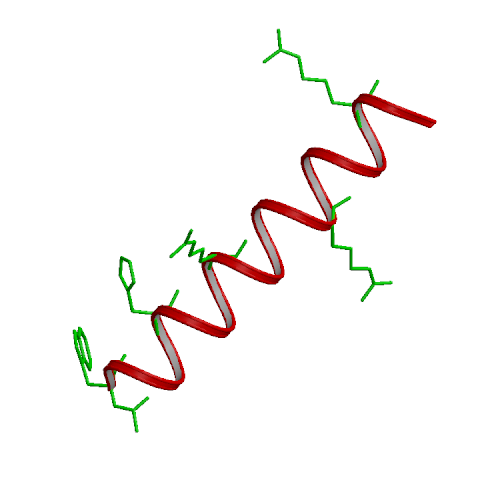
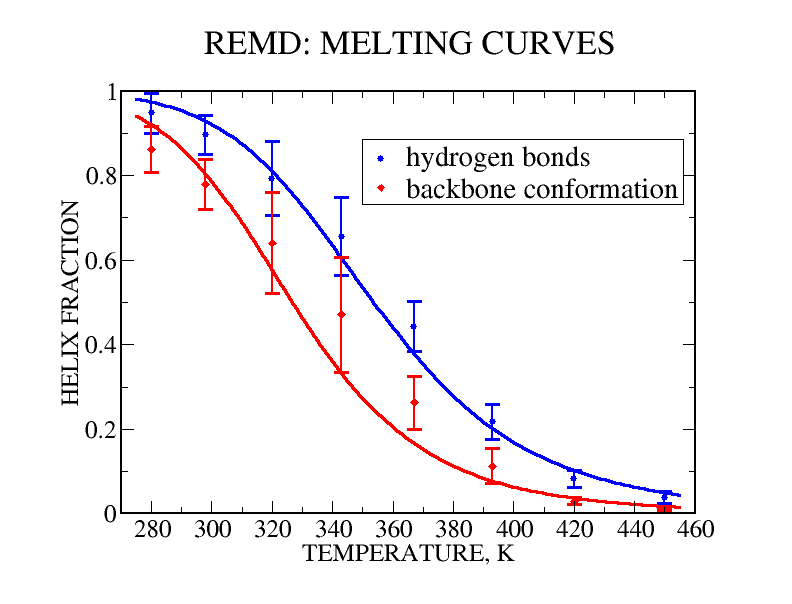
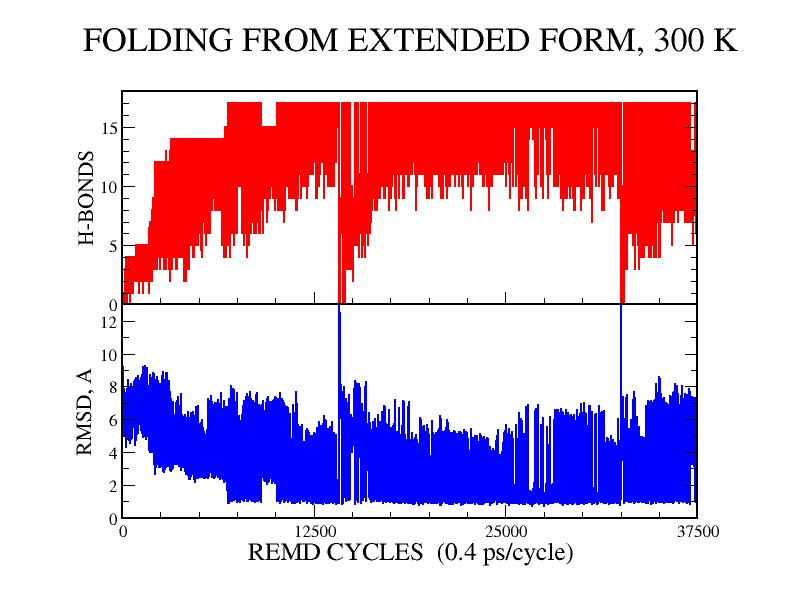
We have performed experimental measurements and computer simulations of the equilibrium structure and folding of a 21-residue alpha-helical heteropeptide. Far UV circular dichroism spectroscopy is used to identify the presence of helical structure and to measure the thermal unfolding curve. The observed melting temperature is 296 K, with a folding enthalpy of -11.6 kcal/mol and entropy of -39.6 cal/(mol K). Our simulations involve 45 ns of replica exchange molecular dynamics of the peptide, using 8 replicas at temperatures between 280 and 450 K, and the program CHARMM with a continuum solvent model. In a 30 ns simulation started from a helical structure, conformational equilibrium at all temperatures was reached after 15 ns. This simulation was used to calculate the peptide melting curve, predicting a folding transition with a melting temperature in the 330-350 K range, enthalpy change of -10 kcal/mol and entropy change of -30 cal/(mol K). The simulation results were also used to analyze the peptide structural fluctuations and the free energy surface of helix unfolding. The fully helical state was the global free energy minimum at low temperatures and the polyproline II helix the major regular conformer above the melting point. In a separate 15 ns replica exchange molecular dynamics simulation started from the extended structure, the helical conformation was first attained after about 2.8 ns, and equilibrium was reached after 10 ns of simulation. These results showed a sequential folding process with a systematic increase in the number of hydrogen bonds until the helical state is reached, and confirmed that the alpha-helical state is the global free energy minimum for the peptide at low temperatures.
|

| Ideal alpha-helix | Ideal PPII helix |
|
| REMD melting curves | Folding simulation |