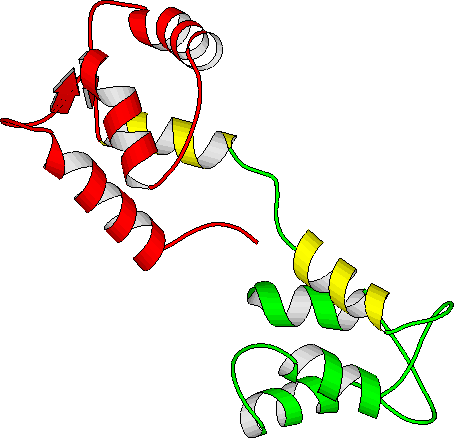
|
|
| Apo-calmodulin NMR solution structure | Apo-calmodulin dynamics animation |
| (PDB file 1CFD) | 4-ns PME trajectory |
_______________________________________________________________________________
C. Yang, and K. Kuczera,
J. Biomol. Struct. Dyn., (2002), 19, 801-820.
A 4-ns molecular dynamics simulation of calcium-free calmodulin in solution has been performed, using Ewald summation to treat electrostatic interactions. Our simulation results were mostly consistent with solution experimental studies, including NMR, fluorescence and x-ray scattering. The secondary structures within the N- and C-terminal domains were conserved in the simulation, with trajectory structures similar to the NMR-derived model structure 1CFD. However, the relative orientations of the domains, for which there are no NMR restraints, differed in details between the simulation and the 1CFD model. The most interesting information provided by the simulations is that the dynamics of calcium-free calmodulin in solution is dominated by slow rigid body reorientations of the domains. The interdomain distance fluctuated between 29 and 39 A, and interdomain orientation angle, defined as the pseudo-dihedral formed by the four calcium binding sites, varied between -2 and 108o. Similarly, the domain linker region also exhibited significant fluctuations, with its length varying in the 34-45 A range and its bend angle in the 10-100o range. The simulations are in accord with fluorescence results suggesting that calcium-free calmodulin is more compact and more flexible than the calcium activated form. Surprisingly, quite similar solvent accessibilities of the hydrophobic patches were seen in the calcium-free trajectory described in this work and previously generated calcium-loaded calmodulin simulations. Thus, our simulations suggest a re-examination of the standard model of the structural change of calmodulin upon calcium binding, involving exposure of the hydrophobic patches to solvent.
|
|
|
| Apo-calmodulin NMR solution structure | Apo-calmodulin dynamics animation |
| (PDB file 1CFD) | 4-ns PME trajectory |
Images and animations from this trajectory are available at our website http://oolung.chem.ukans.edu/~kuczera/1cfd/1cfd.html
_______________________________________________________________________________
J. Mahadevan, C. Xu, T. Siaahan and K. Kuczera
J. Biomol. Struct. Dyn., (2002), 19, 775-788.
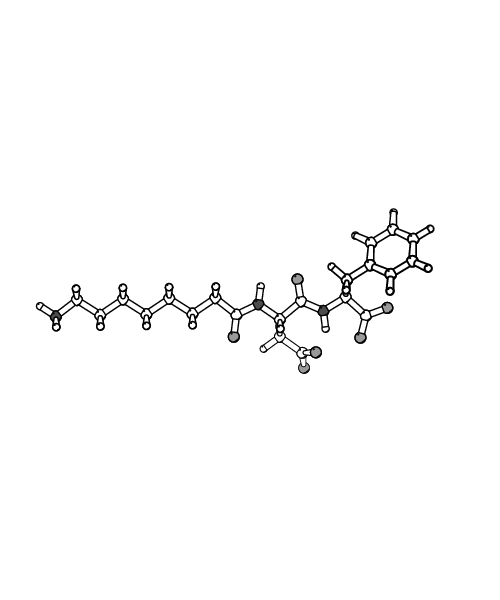
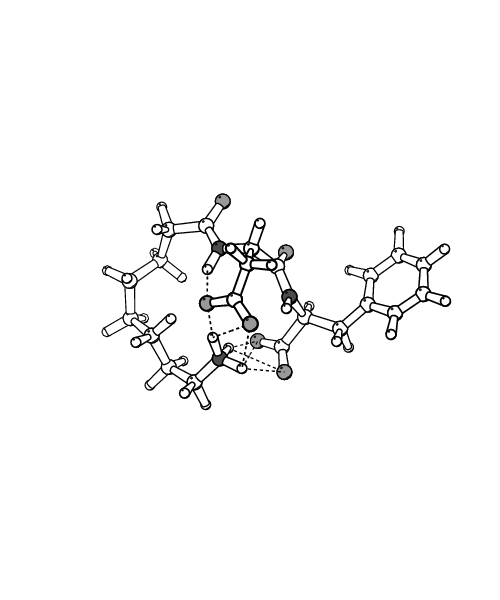
Conformations available to a class of cyclic prodrugs and corresponding linear RGD peptidomimetics were explored using 1 ns length molecular dynamics simulations performed with the program CHARMM. Water and octane, modeled explicitly, were used as solvents to mimic the change of the environment experienced by the solutes upon partition from water to membrane in the trans-cellular transport process. In water, the linear peptidomimetics tended to populate extended-like structures, characterized by strong favorable interactions with solvent and low intrinsic stability. In these extended conformations the charged termini are able to assume large distances, above 15 \AA~ for the longest systems. These linear peptidomimetics have been found to exhibit the highest potency in experimental studies, in accord with the trends experimentally observed for RGD peptides. In contrast, in octane compact conformers of the linear peptidomimetics were favored, with all charged groups aggregated and shielded from solvent, exhibiting high intrinsic stability and weak solute-solvent interactions. Our calculations predict a large unfavorable energy change for transferring the linear systems from water to octane, in agreement with experimental findings that these compounds are not transported via the trans-cellular pathway. The cyclic prodrugs did not exhibit major structural differences between the simulations in water and octane, adopting turn-like conformations in both solvents. The limited response of the cyclic structures to changes in the environment leads to energies of transfer from water to octane that are also unfavorable, but much less so than for the linear molecules. This effect is in accord with the observed enhanced passive trans-cellular transport of the cyclic prodrugs.
|
|
| Extended structure of n=7 system | Compact structure of n=7 system |
| in aqueous solution | in octane |
_______________________________________________________________________________
G. S. Jas and K. Kuczera
Proteins, (2002), 48, 257-268.
In the course of aging or under conditions of oxidative stress, methionine residues of calmodulin undergo oxidation, leading to loss of biological activity of the protein. We have performed free energy simulations of the effects of C-terminal methionine side chain oxidation on the properties of calmodulin. The simulation results indicate that oxidation should have a destabilizing effect on all three protein functional states: calcium free, calcium loaded and with both calcium and target peptide bound. Since the different states are destabilized by different amounts, this leads to a more complex pattern in the observable effects on protein thermal stability, calcium affinity and binding of a target peptide. The influence of oxidation on the free energy of CaM unfolding is estimated by comparing the free energy cost of oxidizing a Met residue in a Gly-Met-Gly peptide and in the protein. The protein thermal stability of the oxidized forms is lowered by a moderate amount 1-3 kcal/mol, in qualitative agreement with experimental results of 0.3 kcal/mol. The calculated changes in affinity for calcium and for the target peptide show opposing trends. Oxidation at position 144 is predicted to enhance peptide binding and weaken calcium binding, while oxidation at 145 weakens peptide binding and enhances affinity for calcium. The lower affinity of Met 145-oxidized calmodulin towards the target peptide correlates with experimentally observed lowering of calmodulin-activated Ca-ATPase activity when oxidized calmodulin from aged rat brains is employed. Thus, our simulations suggest that Met 145 is the oxidation site in the C-terminal fragment of calmodulin. The microscopic mechanism behind the calculated free energy changes appears to be a greater affinity for water of the oxidized Met sidechain relative to normal Met. Structures with Met exposed to solvent had consistently lower free energies than those with buried Met sidechains.

|
| Calcium-loaded calmodulin (1CLL) |
| N-term in red, C-term in blue, central linker in green |
| Methionines in yellow (144 and 145 at bottom of figure) |
_______________________________________________________________________________
I. T. Makagiansar, P. D. Nguyen, A. Ikesue, K. Kuczera, W. Dentler, J. L. Urbauer, N. Galeva, M. Alterman and T. Siahaan
J. Biol. Chem., (2002), 277, 16002-16010.
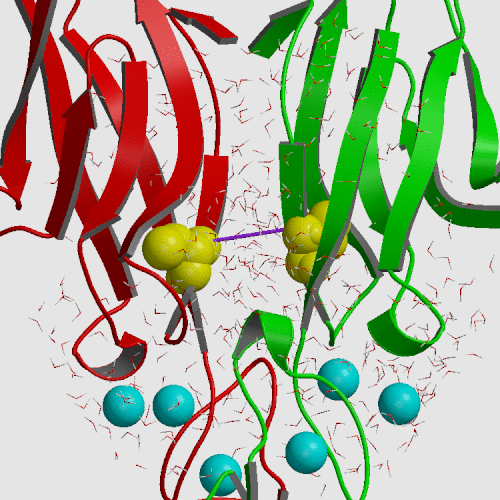
Cadherin is a cell adhesion molecule crucial for epithelial and endothelial cell monolayer integrity. The previously solved x-ray crystallographic structure of the E-CAD12 cis-dimer displayed an upaired Cys9 which protuded away from the Cys9 on the other protomer. To investigate the possible biological function of Cys9 within the first repeat (the E-cadherin-derived N-terminal repeat), E-CAD1, was overexpressed and secreted into the periplasmic space of E. coli cells. Recombinant E-CAD1 produced a mixed monomer and dimer in an equilibrium fashion. The dimer was linked by a disulfide bond through Cys9 pairing. Analysis by high pressure liquid chromatography and electron microscopy suggested the existence of oligomeric complexes. Mutation at Trp2 appears to indicate that these oligomeric complexes trans-dimerize. Interestingly, mutations at Cys9 affected not only the cis-dimerization but also the trans-dimerization of E-CAD1. Accordingly, it is plausible that, under oxidative stress, the homophilic interactions of E-cadherins through E-CAD1 may be promoted and stabilized by this disulfide bond.
Umbrella sampling molecular dynamics was employed to change the S...S distance between Cys9 sulfurs on neighboring protomers from 14.8 A found in the crystal to 5 A, producing a free energy increase of only 1.3 kcal/mol. Subsequent modeling by rotation of chi-1 dihedrals allowed construction of a configuration consistent with a disulfide bond at an S...S distance of 2.1 A. Thus, formation of the S-S bond is structuraly and thermodynamically feasible.
|
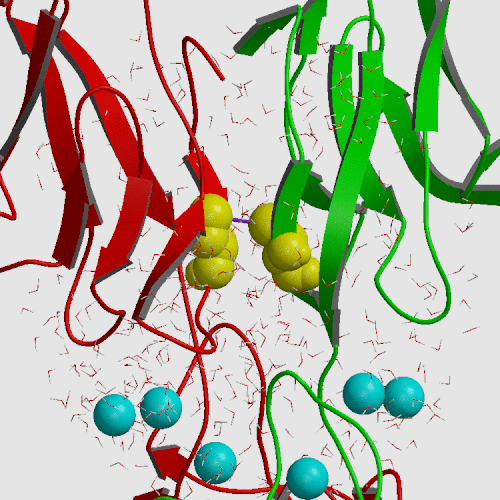
|
| E-cadherin initial crystal structure (PDB file 1FF5) | E-cadherin model structure from umbrella sampling |
| S...S distance 14.8 A | S...S distance 5.0 A |
_______________________________________________________________________________
C. Yang and K. Kuczera
J. Biomol. Struct. Dyn., (2002), 20, 179-197.
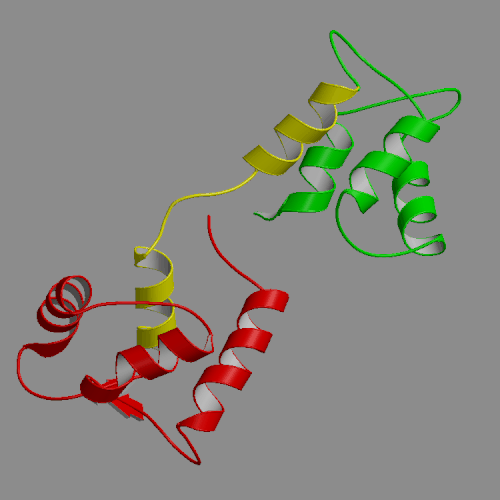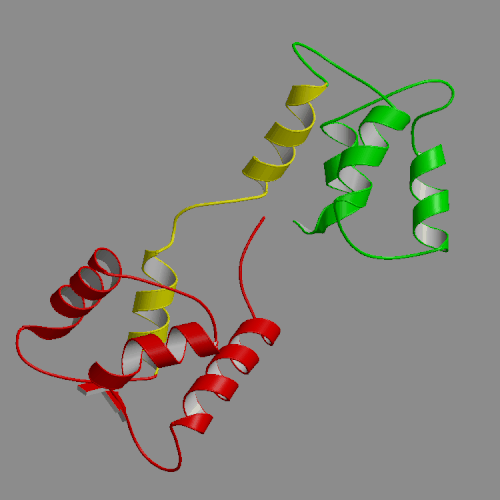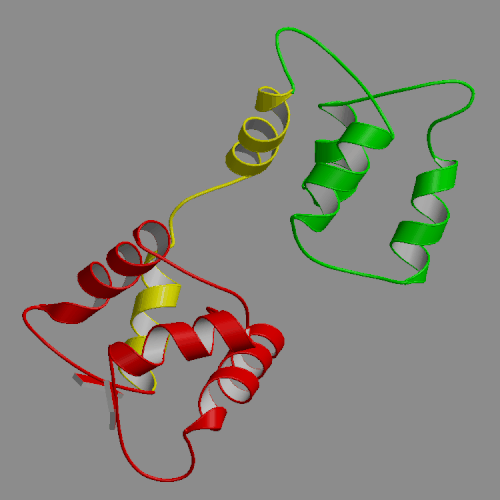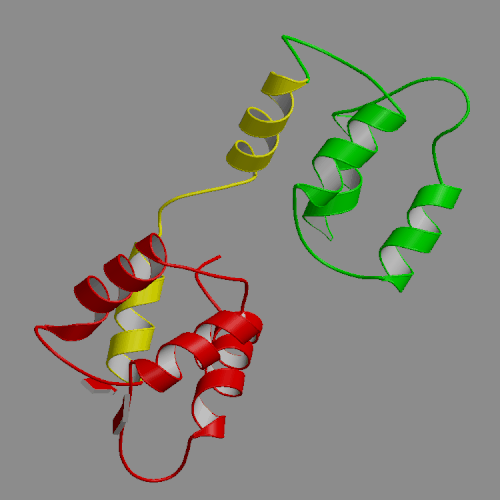
We have performed an 4-ns MD simulation of calmodulin complexed with a target peptide in explicit water, under realistic conditions of constant temperature and pressure, in the presence of a physiological concentration of counterions and using Ewald summation to avoid truncation of long-range electrostatic forces. During the simulation the system tended to perform small fluctuations around a structure similar to, but somewhat looser than the starting crystal structure. The calmodulin-peptide complex was quite rigid and did not exhibit any large amplitude domain motions such as previously seen in apo- and calcium-bound calmodulin. We analyzed the calmodulin-peptide interactions by calculating buried surface areas, CHARMM interaction energies and continuum model interaction free energies. In the trajectory, the protein surface area buried by contact with the peptide is 1373 A2, approximately evenly divided between the calmodulin N-terminal, C-terminal and central linker regions. A majority of this buried surface, 803 A2, comes from nonpolar residues, in contrast to the protein as a whole, for which the surface is made up of mostly polar and charged groups. Our continuum calculations indicate that the largest favorable contribution to peptide binding comes from burial of molecular surface upon complex formation. Electrostatic contributions are favorable but smaller in the trajectory structures, and actually unfavorable for binding in the crystal structure. Since nonpolar groups make up most of buried surface of the protein, our calculations suggest that the hydrophobic effect is the main driving force for binding the helical peptide to calmodulin, consistent with thermodynamic analysis of experimental data. Besides the burial of nonpolar surface area, secondary contributions to peptide binding come from burial of polar surface and electrostatic interactions. In the nonpolar interactions a crucial role is played by the nine methionines of calmodulin. In the electrostatic interactions the negatively charged protein residues and positively charged peptide residues play a dominant role.
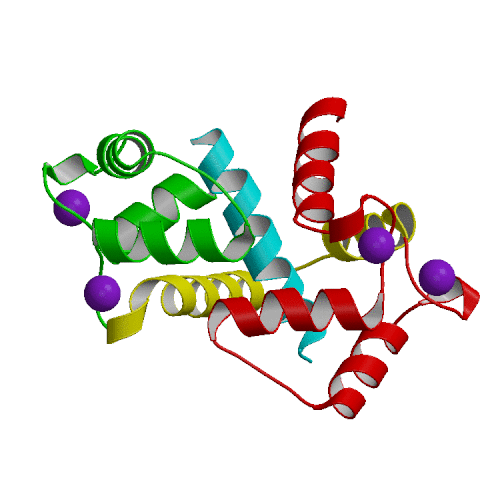
|
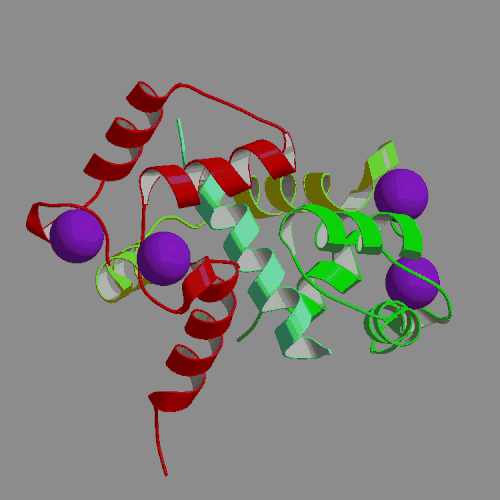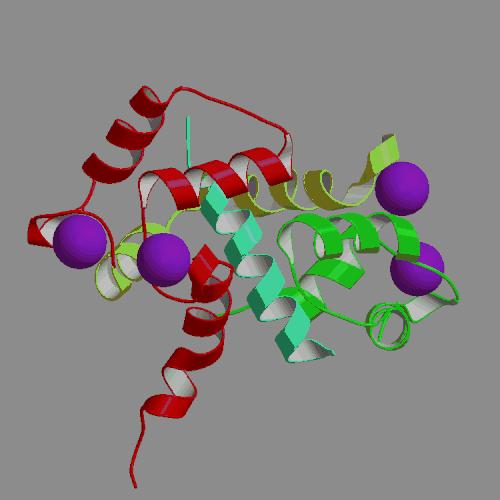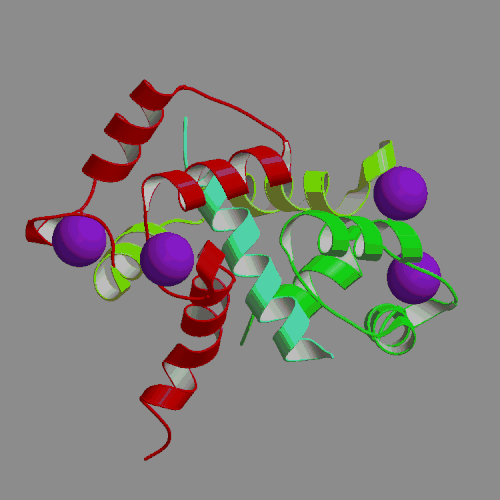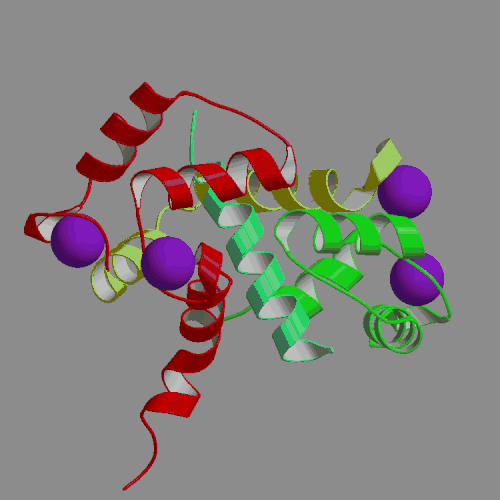
|
| X-ray structure of calmodulin complex | CaM:peptide dynamics animation |
| with smMLCK peptide (PDB file 1CFD) | 4-ns PME trajectory |