|
|
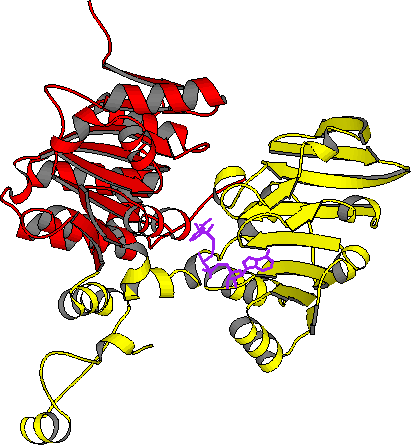
| AdoHcyase: closed form monomer | AdoHcyase: open form monomer |
| PDB file 1A7A; red: cat domain | PDB file 1B3R; yellow: nbd domain |
_______________________________________________________________________________
M. A. Turner, X. Yang, D. Yin, K. Kuczera, R. T. Borchardt and P. L. Howell
Cell Biochem. Biophys., (2001), 33, 101-125.
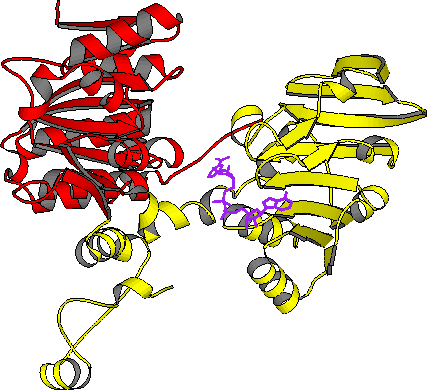
In mammals, S-adenosylhomocysteine hydrolase (AdoHcyase) is the only known enzyme to catalyze the breakdown of S-adenosylhomocysteine (AdoHcy) to homocysteine and adenosine. AdoHcy is the product of all adenosylmethionine (AdoMet)-dependent biological transmethylations. These reactions have a wide range of products, and are common in all facets of biometabolism. As a product inhibitor, elevated levels of AdoHcy suppress AdoMet-dependent transmethylations. Thus, AdoHcyase is a regulator of biological transmethylation in general. The three-dimensional structure of AdoHcyase complexed with reduced nicotinamide adenine dinucleotide phosphate (NADH) and the inhibitor (1'R,2'S,3'R)-9-(2',3'-dihydroxycyclopenten-1-yl)adenine (DHCeA) was solved by a combination of the crystallographic direct methods program, SnB, to determine the selenium atom substructure and by treating the multiwavelength anomalous diffraction data as a special case of isomorphous replacement. The enzyme architecture resembles that observed for NAD-dependent dehydrogenases, with the catalytic domain and cofactor-binding domain each containing a Rossman fold. The two domains form a deep active site cleft containing the cofactor and bound inhibitor molecule. A comparison of the inhibitor complex of the human enzyme and the structure of the rat enzyme, solved without inhibitor, suggests that a 17o rigid bidy movement of the catalytic domain occurs unpon inhibitor/substrate binding.
|
|
|
| AdoHcyase: closed form monomer | AdoHcyase: open form monomer |
| PDB file 1A7A; red: cat domain | PDB file 1B3R; yellow: nbd domain |
J. Mahadevan, K.-H. Lee and K. Kuczera
J. Chem. Phys. B, (2001), 105, 1863-1876.
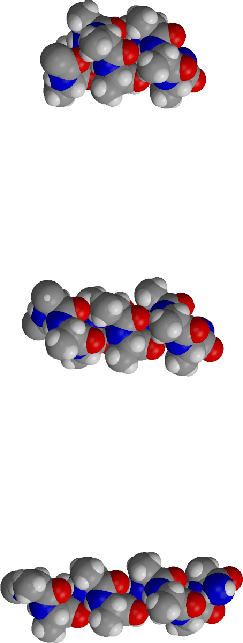
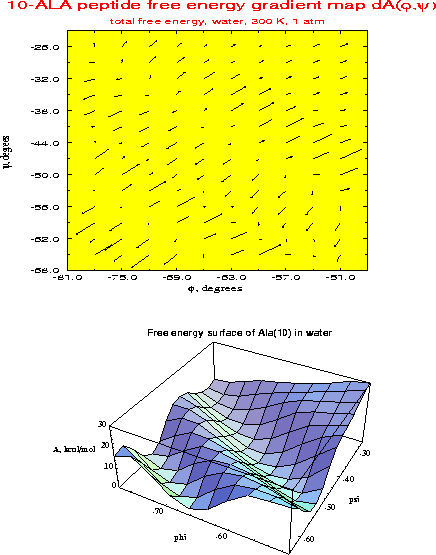
A newly developed molecular simulation algorithm, the multidimensional Conformational Free Energy Thermodynamic Integration (CFTI) method, has been applied to describe conformational free energy surfaces of regular peptide helices in solution. The systems studied are (Ala)10 and (Aib)10 peptides, where Aib is .alpha.-methyl alanine, in water and DMSO solution. The CFTI approach was used to calculate two-dimensional maps of the conformational free energy and its gradient as a function of the peptide backbone dihedrals (.phi.,.psi.). In the region of right-handed helical structures .alpha.-helix and .pi.-helix were found to be locally stable states, corresponding to free energy minima. The location of the minima was refined by free energy optimization. Unexpectedly, solvation by both water and DMSO, tended to strongly stabilize the .pi.-helix relative to the standard .alpha.-helical structure. The .pi. and .alpha. helices had essentially identical stability for (Ala)10 in water and DMSO. The (Ala)10 .pi.-helical free energy minima found in our simulations at (.phi.,.psi.) = (-75o,-56o) in water and at (-78,-53) in DMSO, were markedly different from the generally accepted model values of (-57,-70). Our structure had strong favorable interactions with solvent, low internal strain and a volume identical to that of an .alpha.-helix, suggesting that .pi.-helices should be considered as possible peptide conformers, worthy of further computational and experimental studies. The 310-helix was significantly less stable than the other regular helix types in solution, and no minima corresponding to this form were found in any of the solvated systems, in contrast to previous vacuum simulations for Aib peptides. Energetic and structural features of the different helices were analyzed to provide a microscopic explanation of the stability differences. The large solvation effects and general conformational trends could be rationalized in terms of the interplay between the quality and quantity of intramolecular hydrogen bonds on the one hand and solute-solvent interactions on the other. A relatively inexpensive scheme using vacuum simulations with an approximate solvation correction consisting of a surface term and a Poisson-Boltzmann electrostatic term was able to reproduce the explicit solvent simulation results.
|
|
| ALA10 helices: | ALA10 free energy surface in water showing stable |
| pi, alpha and 310 | alpha- and pi-helix regions |
_______________________________________________________________________________
R. Guenther, H.-J. Hofmann and K. Kuczera,
J. Phys. Chem. B, (2001), 105, 5559-5567.
Hexamers of beta-amino acids with different substituent patterns at the C-alpha and C-beta backbone atoms (none, beta2, beta3, beta2,3, beta3,3, (R,S)beta2,3 and (S,S)beta2,3) were the subject of molecular dynamics simulations on the basis of the CHARMm23.1 force field to generate conformational free energy surfaces for concerted changes in (phi,psi) dihedrals employing a multidimensional conformational integration approach. Application of this technique provides insight into the intrinsic folding propensities of these homooligomers including cooperativity effects. The free energy surfaces provide a complete overview of all possible iperiodic secondary structures. Most striking are the various helix types characterized by hydrogen-bonded pseudocyclces of different size and direction of hydrogen bond formation, e.g. H14, H12 and H10. There are also beta-strand like periodic structures of considerable stability. The formation of various helix types and sheetlike structures strongly depends on the substituent pattern. On the basis of this information it might be possible to design definite secondary structures in beta-peptides to mimic native peptide structures.

Complete free energy surface of beta-peptide hexamer B6
_______________________________________________________________________________
A. Altuve, S. Silchenko, K.-H. Lee, K. Kuczera, S. Terzyan, X. Zhang, D. R. Benson, and M. Rivera,
Biochemistry (2001), 40, 9469-9483.
Two distinct forms of cytochrome b5 exist in the rat hepatocyte. One is associated with the membrane of the endoplasmic reticulum (microsomal, or Mc, cyt b5) while the other is associated with the outer membrane of liver mitochondria (OM cyt b5). Rat OM cyt b5, the only OM cyt b5 identified so far, has a significantly more negative reduction potential and is substantially more stable toward chemical and thermal denaturation than Mc cytochromes b5. In addition, hemin is kinetically trapped in rat OM cyt b5 but not in the Mc proteins. As a result, no transfer of hemin from rat OM cyt b5 to apomyoglobin is observed at pH values as low as 5.2, nor can the thermodyamically favored ratio of hemin orientational isomers be achieved under physiologically relevant conditions. These differences are striking given the similarity of the respective protein folds. A combined theoretical and experimental study has been conducted in order to probe the structural basis behind the remarkably different properties of rat OM and Mc cytochromes b5. Molecular dynamics (MD) simulations starting from the crystal structure of bovine Mc cyt b5 revealed a conformational change that exposes several internal residues to the aqueous environment. The new conformation is equivalent to the "cleft-opened" intermediate observed in a previously reported MD simulation of bovine Mc cyt b5 [Storch, E. M., and Daggett, V. (1995) Biochemistry 34, 9682-9693]. The rat OM protein does not adopt a comparable conformation in MD simulations, thus restricting access of water to the protein interior. Subsequent comparisons of the protein sequences and structures suggested that an extended hydrophobic network encompassing the side chains of Ala-18, Ile-32, Leu-36, and Leu-47 might contribute to the inability of rat OM cyt b5 to adopt the cleft-opened conformation and, hence, stabilize its fold relative to the Mc isoforms. A corresponding network is not present in bovine Mc cyt b5 because positions 18, 32, and 47, are occupied by Ser, Leu, and Arg, respectively. To probe the roles played by Ala-18, Ile-32, and Leu-47 in endowing rat OM cyt b5 with its unusual structural properties, we have replaced them with the corresponding residues in bovine Mc cyt b5. Hence, the I32L (single), A18S/L47R (double), and A18S/L47R/I32L (triple) mutants of rat OM cyt b5 were prepared. The stability of these proteins was found to decrease in the following order: WT rat OM > rat OM I32L > rat OM A18S/L47R > rat OM A18S/L47R/I32L > bovine Mc cyt b5. The decrease in stability of the rat OM protein correlates with the extent to which the hydrophobic cluster involving the side chains of residues 18, 32, 36, and 47 has been disrupted. Complete disruption of the hydrophobic network in the triple mutant is confirmed in a 2.0 A resolution crystal structure of the protein. Disruption of the hydrophobic network also facilitates hemin loss at pH 5.2 for the double and triple mutants, with the less stable triple mutant exhibiting the greater rate of hemin transfer to apomyoglobin. Finally, 1H NMR spectroscopy and side-by-side comparisons of the crystal structures of bovine Mc, rat OM, and rat OM A18S/L47R/I32L cyt b5 allowed us to conclude that the nature of residue 32 plays a key role in controlling the relative stability of hemin orientational isomers A and B in rat OM cyt b5. A similar analysis led to the conclusion that Leu-70 and Ser-71 play a pivotal role in stabilizing isomer A relative to isomer B in Mc cytochromes b5.

|

|
| 1 ns dynamics of rat OM cyt b5 (cleft closed). Heme red, Ala 18 green, Ile 32 cyan, Leu 36 brown, Leu 47 blue. | 1 ns dynamics of bovine Mc cyt b5 (cleft opened) Heme red, Ser 18 green, Leu 32 cyan, Leu 36 brown, Arg 47 blue. |
_______________________________________________________________________________
C. Yang, G. S. Jas and K. Kuczera,
J. Biomol. Struct. Dyn., (2001), 19 247-271.
Two 4-ns molecular dynamics simulations of calcium loaded calmodulin in solution have been performed, using both standard nonbonded cutoffs and Ewald summation to treat electrostatic interactions. Our simulation results are generally consistent with solution experimental studies of calmodulin structure and dynamics, including NMR, cross-linking, fluorescence and x-ray scattering. The most interesting result of the molecular dynamics simulations is the detection of large-scale structural fluctuations of calmodulin in solution. The globular N- and C-terminal domains tend to move approximately like rigid bodies, with fluctuations of interdomain distances within a 7 A range and of interdomain angles by up to 60 deg. Essential dynamics analysis indicates that the three dominant types of motion involve bending of the central helix in two perpendicular planes and a twist in which the domains rotate in opposite directions around the central helix. In the more realistic Ewald trajectory the protein backbone remains mostly within a 2-3 A root-mean-square distance from the crystal structure, the secondary structure within the domains is conserved and middle part of the central helix becomes disordered. The central helix itself exhibits limited fluctuations, with its bend angle exploring the 0-50 deg range and the end-to-end distance falling in 39-43 A. The results of the two simulations were similar in many respects. However, the cutoff trajectory exhibited a larger deviation from the crystal, loss of several helical hydrogen bonds in the N-terminal domain and lack of structural disorder in the central helix.
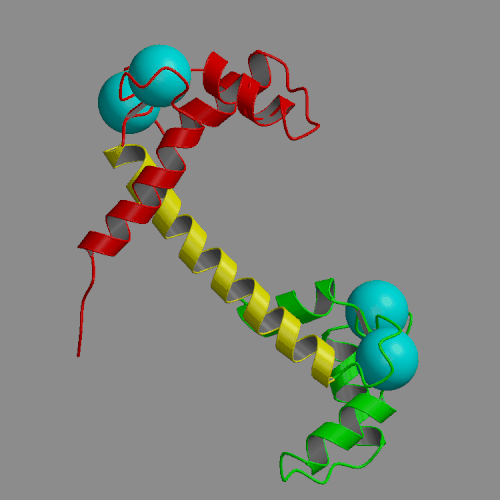
|
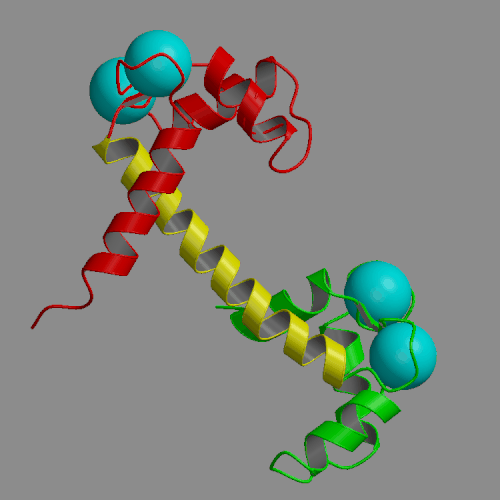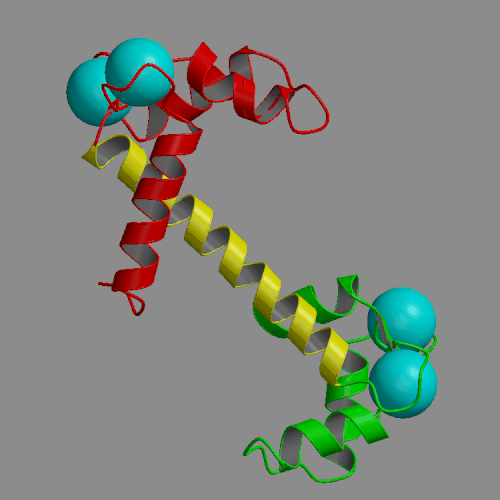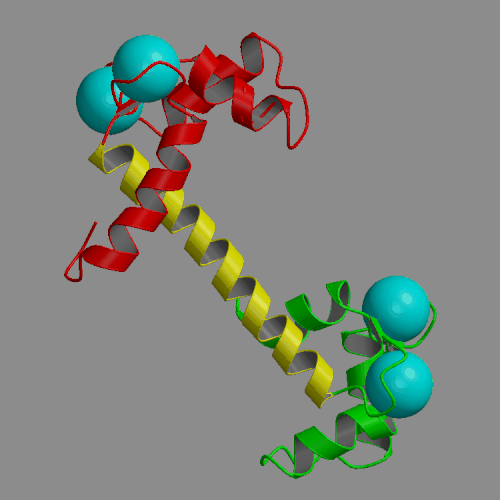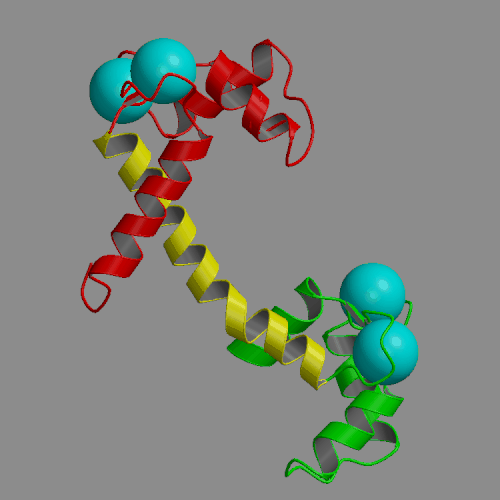
|
| Crystal structure of calcium loaded calmodulin (1CLL) N-terminal domain: red; central helix: yellow; C-terminal domain: green; calcium ions: cyan spheres. | Animation of 4-ns MD trajectory of calcium-loaded calmodulin (with counterions and Ewald summation) |
_______________________________________________________________________________
Y. Hu, X. Yang, D. H. Yin, J. Mahadevan, K. Kuczera, R. L. Schowen and R. T. Borchardt
Biochemistry, (2001), 40 15143-15152.
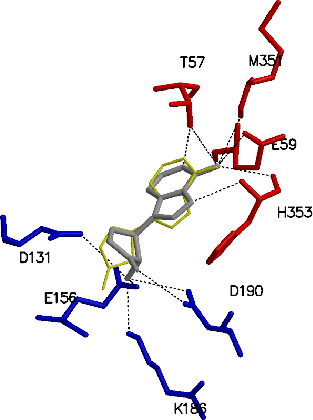
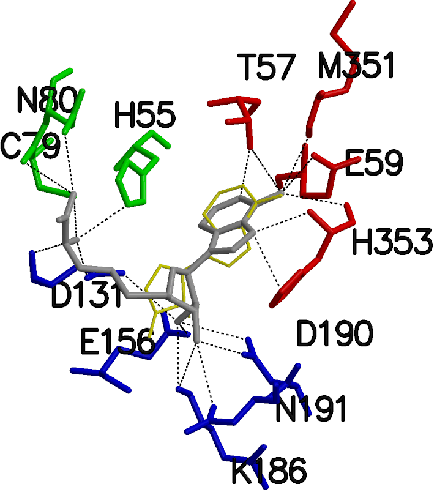
S-Adenosylhomocysteine (AdoHcy) hydrolase catalyzes the reversible hydrolysis of AdoHcy to adenosine (Ado) and homocysteine (Hcy), playing an essential role in modulating cellular Hcy levels and regulating activities of a host of methyltransferases in eukariotic cells. This enzyme exists in an open conformation (active site unoccupied) and a closed conformation (active site occupied with substrate or inhibitor). To investigate the binding of natural substrates during catalysis, the computational docking program AutoDock (with confirming calculations using CHARMM) was used to predict binding modes of various substrates and inhibitors to the closed and open forms of the hydrolase. The results revealed that the interaction between the substrate and the open form of the enzyme is nonspecific, whereas binding of the substrate to the closed form is highly specific with the adenine moiety of a substrate as the main recognition factor. Residues Thr57, Glu59, Glu156, Gln181, Lys186, Asp190, Met351 and His55 are involved in substrate binding, which is consistent with the crystal structure. His 55 in the docked model appears to participate in the elimination of water from Ado through the interaction with the 5'-OH group of Ado. In the same reaction, Asp131 removes a proton from the 4' position of the substrate after the oxidation-reduction reaction of the enzyme. To identify the residues that bind the Hcy moiety, AdoHcy was docked to the closed form of AdoHcy hydrolase. The Hcy tail is predicted to interact with His55, Cys79, Asn80, Asp131, Asp134 and Leu344 in a strained conformation, which may lower the reaction barrier and enhance the catalysis rate.
|
|
| CHARMM docking of inhibitor 3'-keto-DHCeA (grey) vs. crystal structure (gold) in active site of closed form of AdoHcyase (1A7A) | CHARMM docking of substrate AdoHcy (grey) vs. crystal position of inhibitor 3'-keto-DHCeA (gold) in active site of closed form of AdoHcyase (1A7A) |