D. Yin, K. Kuczera and T. C. Squier
Chem. Res. Toxicol., (2000), 13 , 103-110.
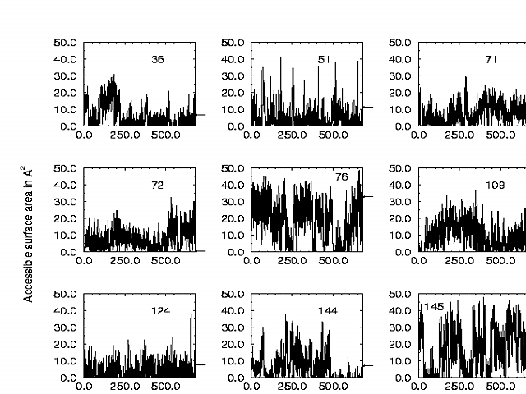
The oxidative modification of methionines within the primary sequence of calmodulin (CaM) results in an inability to activate the PM-Ca-ATPase fully, and may contribute to alterations of calcium homeostasis under conditions of oxidative stress. To identify differences in the sensitivities of CaM isoforms to oxidative modification, we have compared function and patterns of oxidative modification resulting from the exposure of CaM isolated from bovine testes and wheat germ to H2O2. In comparison to CaM isolated from wheat germ, vertebrate CaM is functionally resistant to oxidant-reduced loss of function. The decreased functional sensitivity of vertebrate CaM correlates with at 75 % reduction in the rate of oxidative modification of a methionine near the carboxyl terminus (i.e. Met 144 or Met 145). The extent of oxidative modification to other methionines in these CaM isoforms is similar. These results suggest that the sensitivity of Met 144 or Met 145 to oxidation modulates the ability of CaM to activate the PM-Ca-ATPase. Consistent with this interpretation, a CaM mutant in which glutamines were substituted for Met 144 and Met 145 fully activats the PM-Ca-ATPase irrespective of the oxidative modification of the other seven methionines to their corresponding methionine sulfoxides. The extent of oxidative modification to individual methionines in vertebrate CaM by H2O2 correlates with the time-averaged surface accessibility of individual sulfurs calculated from molecular dynamics simulations. Thus, the sensitivity of individual methionines to oxidative modification is directly related to the solvent accessibility. These results indicate that sequence differences between vertebrate and plant CaM alter the sensitivity of methionines near the carboxyl terminus to oxidative modification because of alterations in their solvent accessibility. We suggest that these sequence differences between CaM Isoforms have a regulatory role in modulating the functional sensitivity of CaM to conditions of oxidative stress.
_______________________________________________________________________________
E. V. Rybak-Akimova and K. Kuczera,
Inorg. Chem., (2000), 39, 2462-2472.
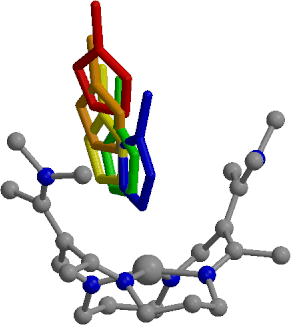
Cobalt(II) complexes with tetradentate macrocyclic cyclidene ligands are known to coordinate one additional axial base molecule, leaving the sixth vacant coordination site at the metal available for small ligand (e.g. O2) binding. Molecular mechanics and molecular dynamics simulations provide a microscopic view of 1-methylimidazole (MeIm) binding within the cavities of several lacunar (bridged) and saddle-shaped (unbridged) cyclidenes and uncover the roles of the bridges and the walls of the clefts in steric protection of the cobalt(II) coordination site. Short bridges (C3 and C6) prevent inside-the-cavity MeIm binding because of severe ligand distortions leading to high energy penalties (58 and 25 kcal/mol, repsectively), while long bridges (C8 and C12) flip away from the MeIm binding site, allowing for penalty-free MeIm inclusion. In the unbridged saddle-shaped complex there is no difference between inside- and outside-the-cavity MeiM binding. The preferential existence of the coordinatively unsaturated, five-coordinate species Co(unbrCyc)(MeIm)2+ should therefore be explained by electronic, rather than steric, factors. Molecular dynamics and free energy simulations reveal the presence of a weak (ca. 4 kcal/mol in the gas phase and ca. 2 kcal/mol in methanol solution) noncovalent MeIm binding site at the entrance of the cleft of Co(II) unbridged cyclidene, at a distance of about 4 A from the metal ion. The macrocycle remains undistorted at such large Co-N(MeIm) separations, while the cavity opens up by 0.9 A upon covalent MeImbinding (Co-N(MeIm) distance of 2 A). An increase in macrocyclic strain energy upon MeIm inclusion is compensated by favorable nonbonded interactions with the incoming base and the walls of the unbridged cyclidene.
|
_______________________________________________________________________________
D. Yin, X. Yang, Y. Hu, K. Kuczera, R. L. Schowen, R. T. Borchardt and T. C. Squier,
Biochemistry (2000), 39, 9811-9818.
Comparison of the crystal structures of S-adenosylhomocysteine (AdoHcy) hydrolase in the substrate free, NAD+ form [Hu et al., (1999) Biochemistry , 38, 8323-8333], and substrate-bound, NADH form [ turner et al. (1998) Nat. Struct. Biol. 5, 369-376], indicates large differences in the spatial arrangements of the catalytic and NAD+ binding domains. The substrate free, NAD+ form exists in an "open" form with respect to the catalytic and NAD+ binding domains, whereas the substrate-bound, NADH form exists in a closed form with respect to those domains. To address whether domain closure is induced by substrate binding or its subsequent oxidation, we have measured the rotational dynamics of spectroscopic probes covalently bound to Cys113 and Cys421 within the catalytic and carboxyl-terminal domains. An independent domain motion is associated with the catalytic domain prior to substate binding, suggesting the presence of a flexible hinge element between the catalytic and NAD+ binding domains. Following binding of substrates (i.e. adenosine or neplanocin A) or a nonsubstrate (i.e. 3'-deoxyadenosine), the indpendent domain motion associated with the catalytic domain is essentially abolished. Likewise, there is a substantial decrease in the average hydrodynamic volume of the protein that is consistent with a reduction in the overall dimensions of the homotetrameric enzyme following substrate binding and oxidation observed in earlier crystallographic studies. Thus, the catalytic and NAD+ binding domains are stabilized to form a closed active site through interactions with the substrate prior to substrate oxidation.

|

|
| CLOSED FORM | OPEN FORM |
_______________________________________________________________________________
C. E. MacBeth, A. P. Golombek, V. G. Young Jr., C. Yang, K. Kuczera, M. P. Hendrich and A. S. Borovik,
Science , (2000), 289, 938-941.
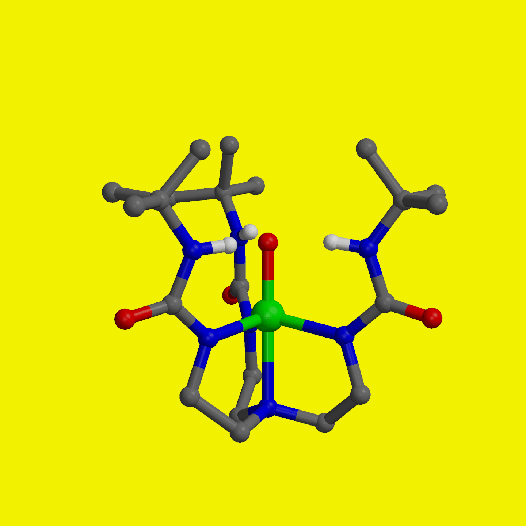
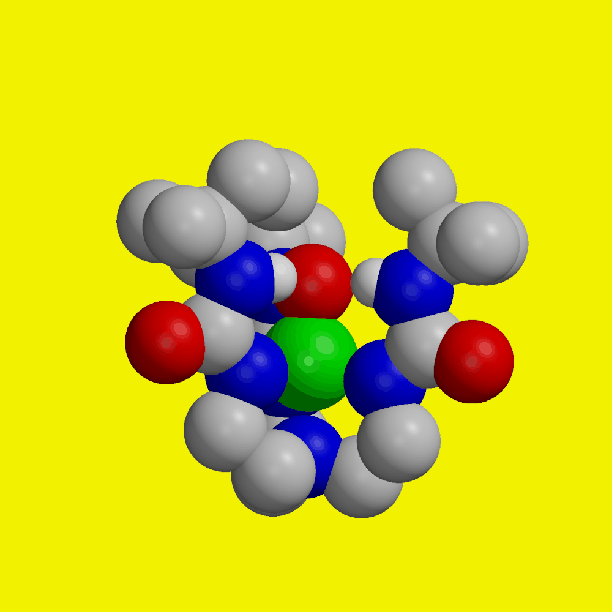
Iron species with a terminal oxo ligand are implicated as key intermediates in several synthetic and biochemical catalytic cycles. However, there is a dearth of structural information regarding these types of complexes because their instability has precluded isolation under ambient conditions. The isolation and structural characterization of an iron(III) complex with a terminal oxo ligand, derived directly from oxygen (O2), is reported. A stable structure resulted from placing the oxoiron unit within a synthetic cavity lined with hydrogen-bonding groups. The cavity creates a microenvironment around the iron center that aids in regulating O2 activation and stabilizing the oxoiron unit. These cavities share properties with the active sites of metalloproteins, where function is correlated strongly with site structure.
Isolation of these complexes is achieved using the hydrogen-bonding tripodal ligand tris[(N'-tert-butylureaylato)-N-ethyl)]aminato ([H31]3-). The v(FeO) band in [Fe(III)[H31(O)]2- is observed at 671 cm-1. 18O-labeling studies confirm that the oxo ligand in the Fe(III)-oxo complex [Fe(III)[H31(O)]2- and the hydroxo oxygen atom in the Fe(III)-OH complex [Fe(III)[H31(OH)]1- are derived from O2. Spectroscopic and X-ray diffraction studies on [Fe(III)[H31(O)]2-, [Fe(III)[H31(OH)]1- and [Fe(II)[H31(OH)]2- are consistent with each complex having a trigonal bipyramidal coordination geometry. The Fe(III)--O distance in [Fe(III)[H31(O)]2- is 1.813(3) A. In [Fe(III)[H31(O)]2- three intramolecular hydrogen bonds exist between the oxo group and the N-H groups of the urea tripodal ligand. EPR and Mossbauer measurements show that the [Fe(III)[H31(O)]2- and [Fe(III)[H31(OH)]1- have S=5/2 ground states.
|
|
_______________________________________________________________________________
G. S. Jas, E. Larson, C. K. Johnson, and K. Kuczera,
J. Phys. Chem. A, (2000), 104, 9841-9852.
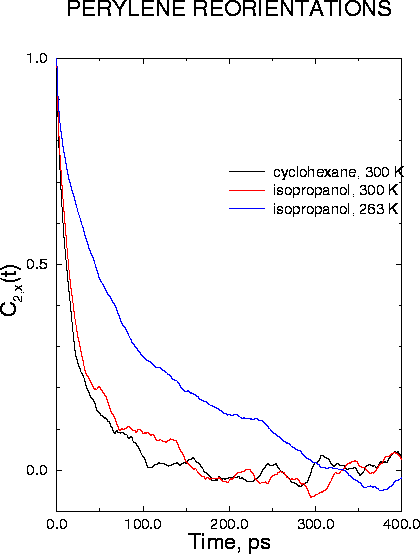
Molecular dynamics simulations and time resolved linear dichroism measurements have been employed to investigate rotational diffusion of perylene in two organic solvents, cyclohexane, a non-polar solvent, and 2-propanol, a polar solvent. Both experiments and simulations yield a bi-exponential rotational anisotropy decay for the long in-plane axis. The calculated time constants were 9 ps and 44 ps in cyclohexane at 300 K, 13 ps and 75 ps in 2-propanol at 300 K, and 25 ps and 126 ps in 2-propanol at 263 K, in excellent agreement with corresponding time-resolved linear dichroism measurements of 10 ps and 40 ps, 12 ps and 70 ps, and 23 ps and 175 ps respectively. Although the viscosity of 2-propanol is more than two times that of cyclohexane at room temperature, the measured rotational reorientation times and the calculated average rotational diffusion coefficients of perylene are similar in the two solvents, demonstrating a breakdown of simple hydrodynamic theory. Analysis of the calculated rotational diffusion coefficients for the individual molecular axes showed that diffusion was highly anisotropic, with the fastest rotation around the out-of-plane axis z. This dominant motion occurred at comparable rates for perylene in cyclohexane and 2-propanol, leading to similar values of average rotational diffusion coefficients in the two solvents. The hindered spinning of perylene in cyclohexane relative to 2-propanol could be rationalized in terms of tighter packing of the former solvent around the solute in the molecular plane.

|
|
_______________________________________________________________________________
K.-H. Lee, D. R. Benson and K. Kuczera,
Biochemistry, (2000), 39, 13737-13747.
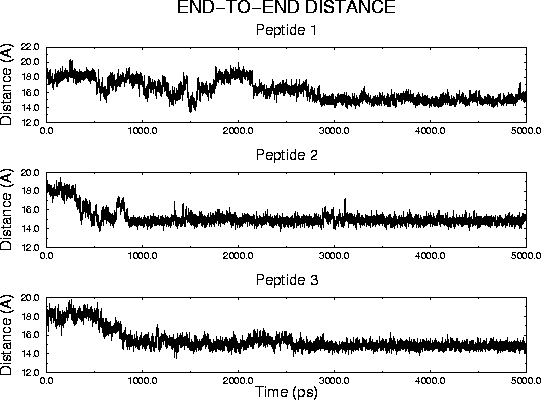
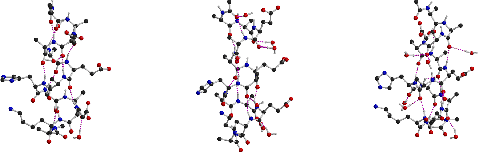
Molecular dynamics simulations were carried out for 13-residue peptides of the form AcNH-A-A-E-X-A-E-A-H-A-A-E-K-A-CONH2 with X = A, F and W. All three peptides exhibited unexpected dynamical behavior, undergoing a transition from an .alpha.-helical to a .pi.-helical structure in the course of 5-ns trajectories in aqueous solution. Analysis of peptide length, accessible surface, interaction energies, hydrogen bonding and dihedral angles was consistent with .alpha. to .pi. transitions at 2800 ps, 500 ps and 800 ps for X = A, F and W, respectively. The transitions occurred sequentially and cooperatively, propagating from the C- to the N-terminus for X = A and W, and from the center towards both termini for X = F. The time scale of the overall transition ranged from 300 to 500 ps. For all three peptides the backbone structural transition was accompanied by a concerted rearrangement of the charged sidechains, including a 3 A increase in the distance between carboxylate groups of Glu 3 and Glu 6. During the transition the peptide backbone hydrogen bonding patterns were disrupted at the interface between the .alpha.-helical and nascent .pi.-helical regions, with peptide groups forming water-bridged hydrogen bonds. The peptide structures exhibited significant fluidity, with individual residues sampling .alpha.-, .pi.- and 310-helical conformations, as well as a "coil" state, without any intramolecular hydrogen bonds. The studied peptides have been designed to form .alpha.-helices when incorporated in novel hemeprotein model compounds, peptide-sandwiched-mesohemes, which consist of two identical peptides covalently attached to a Fe(III) mesoporphyrin (Liu et al., J. Am. Chem. Soc., 1999, 121:11798-11812). The possibility of adopting .pi.-helical structures by the constituent peptides may influence the properties of the hemeprotein models.
|
|
| End-to-end distance evolution | Peptide 1 : structures along the transition path |
| for peptides 1, 2 and 3 | left: alpha-helix; center: intermediate; right: pi-helix |
_______________________________________________________________________________