|

|
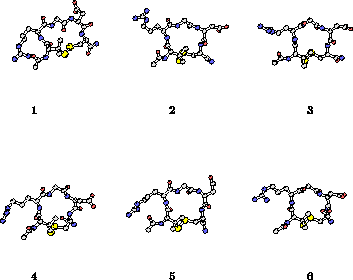
| Sample of cyclic RGD peptide structures | RMS deviations between structures from the two linear RGD trajectories: yellow: backbone RMSD below 1 A, purple: above 3 A. |
Y. Wang, S.-Y. Goh and K. Kuczera
J. Peptide Res., (1999), 53 , 188-200.
Three 1 ns length molecular dynamics simulations of an RGD peptide (Ac-Pen-Arg-Gly-Asp-Cys-NH2, with Pen denoting penicillamine) have been performed in aqueous solution, one for the disulfide bridged, and two for the unbridged form. The trajectories were analyzed to identify conformations explored by the two forms and to calculate several properties - NMR vicinal coupling constants, order parameters, dipole moments and diffusion coefficients, in an effort to describe the physical role of the disulfide bond. The cyclic peptide was able to explore several distinct backbone conformations centered around a turn-extended-turn structure. However, its flexibility was limited and it appeared to be "locked in" into a a family of structures characterized by a high dipole moment and a well defined conformation of the pharmacophore, which has been previously identified as biologically active. Excellent agreement between the simulated and observed NMR vicinal coupling constants indicates that realistic structures were sampled in the cyclic peptide simulation. The linear form of the peptide was much more flexible than the cyclic one. In the two independent 1 ns simulations of the linear form the explored conformations could be roughly grouped into two classes, of cyclic-like and extended type. Within each simulation the peptide switched between the two classes of structures several times. Exact matches between conformations in the two linear peptide simulations were not found, but several conformational regions with backbone rms deviations below 1 A were identified, suggesting that representative structures of the linear form have also been identified. In the linear peptide simulations the RGD pharmacophore is able to adopt a wide range of conformations, including the one preferred by the cyclic form. The lower biological activity of the linear peptide as compared to the cyclic one may be correlated with the lower population of this structure in the absence of the disulfide bond.
|
|
|
| Sample of cyclic RGD peptide structures | RMS deviations between structures from the two linear RGD trajectories: yellow: backbone RMSD below 1 A, purple: above 3 A. |
_______________________________________________________________________________
Y. Wang and K. Kuczera
Theor. Chem. Acc. , (1999), 101 , 274-281.
We have calculated the free energy differences between four conformers of the linear form of the opioid pentapeptide DPDPE in aqueous solution. The conformers are Cyc, representing the structure adopted by the linear peptide prior to disulfide bond formation, beta-C and beta-E, two slightly different beta-turns previously identified in unconstrained molecular dynamics simulations, and Ext, an extended structure. Our simulations indicate that beta-E is the most stable of the studied conformers of linear DPDPE in aqueous solution, with beta-C, Cyc and Ext having free energies higher by 2.3, 6.3, and 28.2 kcal/mol, respectively. The free energy differences of 4.0 kcal/mol between beta-C and Cyc, and 6.3 kcal/mol between beta-E and Cyc, reflect the cost of pre-organizing the linear peptide into a conformation conducive for disulfide bond formation. Such a conformational change is a pre-requisite for the chemical reaction of S--S bond formation to proceed. The relatively low population of the cyclic-like structure agrees qualitatively with observed lower potency and different receptor specificity of the linear form relative to the cyclic peptide, and with previous unconstrained simulation results. Free energy component analysis indicates that the moderate stability difference of 4.0-6.3 kcal/mol between the beta-turns and the cyclic-like structure results from cancellation of two large opposing effects. In accord with intuition, the relaxed beta-turns have conformational strain 43-45 kcal/mol lower than the Cyc structure. However, the cyclic-like conformer interacts with water about 39 kcal/mol strongly than the open beta-turns. Our simulations are the first application of the recently developed multidimensional Conformational Free energy Thermodynamic Integration (CFTI) protocol to a solvated system, with fast convergence of the free energy obtained by fixing all flexible dihedrals. Additionally, the availability of the CFTI multidimensional free energy gradient leads to a new decomposition scheme, giving the contribution of each fixed dihedral to the overall free energy change and providing additional insight into the microscopic mechanisms of the studied processes.
| 
| 
| 
|
| Cyc | Ext | beta-C | beta-E |
_______________________________________________________________________________
E. V. Rybak-Akimova, K. Kuczera, G. S. Jas, Y. Deng and D. H. Busch
Inorg. Chem., (1999), 38 , 3423-3434.
Molecular dynamics simulations have been used to study the three-dimensional distribution of methanol solvent molecules around three cobalt(II) cyclidene complexes differing in details of their ligand structures. The ligands are a planar unbridged 14-membered macrocycle in Co([14]Cyc), a saddle-shaped unbridged 16-membered macrocycle in Co([16]Cyc), and a lacunar bridged 16-membered macrocyle in Co(C6[16]Cyc). All three complexes contain five-coordinate cobalt(II) with the metal ion bound to four nitrogen donor atoms from the macrocycle and one nitrogen donor from an axial methylimidazole. Distinctly different solvation patterns are exemplified for the three complexes by the position of the maximum in the Co--O pair distribution function (at r(Co--O) = 2.5, 3.7 and 4.5 A for Co([14]Cyc), Co([16]Cyc) and Co(C6[16]Cyc), respectively) and by the average number of methanol molecules within the macrocyclic cleft (1.5, 0.7 and 0.4 molecules within a Co--O distance of 5.25 A in the "cavity", respectively). Analysis of the anisotropic solvent structure reveals the presence of a methanol molecule directly above the cobalt(II) center, at a distance of ca. 2.5 A for planar Co([14]Cyc) and the absence of solvent from the close proximity of the metal for the remaining complexes. The bridge further protects the sides of the cavity from the solvent. The width of an empty cavity of Co(C6[16]cyc) shrinks by 0.3 A in methanol solution as compared to vacuum simulations. These results confirm the experiemntally based (decrease in absolute value of entropies and enthalpies of oxygen binding) suggestion that extensive solvation of the cobalt(II) center reduces its accessibility to incoming small molecules.
_______________________________________________________________________________
J. Wiórkiewicz-Kuczera, K. Kuczera, C. Bazzicalupi, A. Bencini, B. Valtancoli, A. Bianchi and K. Bowman-James
New J. Chem., (1999), 23 , 1007-1013.
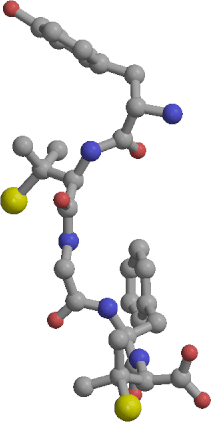
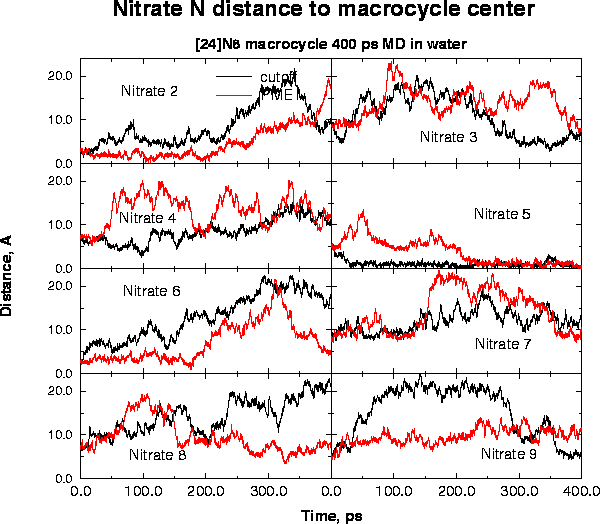
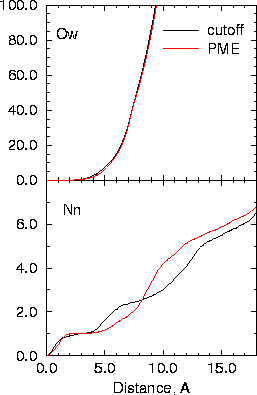
The crystal structure of a hexaprotonated octaaza macrocycle 1,4,7,10,13,16,19,21-octaazacyclotetracosane, has been obtained as the nitrate complex, H81.8NO3. The complex crystallizes in the monoclinic space group P21/c with unit cell parameters a = 14.43(2), b = 8.36(1), c = 15.9(1) A, beta = 110.8(2) deg and U = 1793(12) A3. The macrocycle crystallizes in a relatively flat elliptical conformation with dimensions of 11.9 and 7.2 A and a depth of 1.8 A. The crystallographic coordinates formed the basis of molecular dynamics simulations of the macrocycle and nitrate counterions in aqueous solution over a 400 ps timeframe. The major conformational change compared to the crystal was the the shifting of all NCCN dihedral angles to the trans conformation in the simulations. Also, conformational transitions in CNCC and CCNC angles flanking a central CC bond were found to be correlated. The macrocycle remained relatively flat during the simulations, resulting in essentially complete hydration during the first 100 ps of the simulation. Extensive water relay networks could be identified throughout the simulation, which are attributed to maintaining and stabilizing anion/cation interactions in solution.
|
|
|
| macrocycle crystal structure showing all NH2+ groups | Time course of nitrate-macrocycle distances | Average number of waters and nitrates within a sphere around macrocycle |
_______________________________________________________________________________
{kind=link}
{kind=link}
{kind=link}