
|
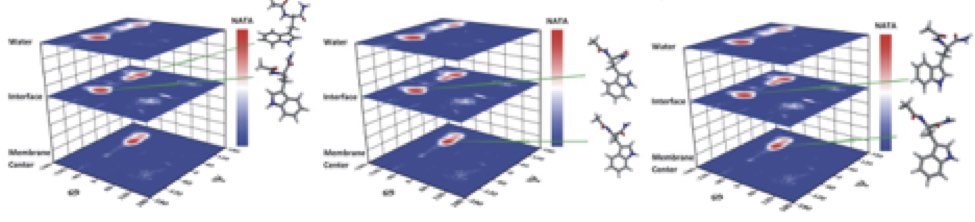
| Snapshot of 2x25 DOPC bilayer. | NATA dipeptide conformations in DOPC bilayer. |
_______________________________________________________________________________
G.S. Jas and K. Kuczera
J. Phys. Chem. B, 122:10806-10816 (2018)
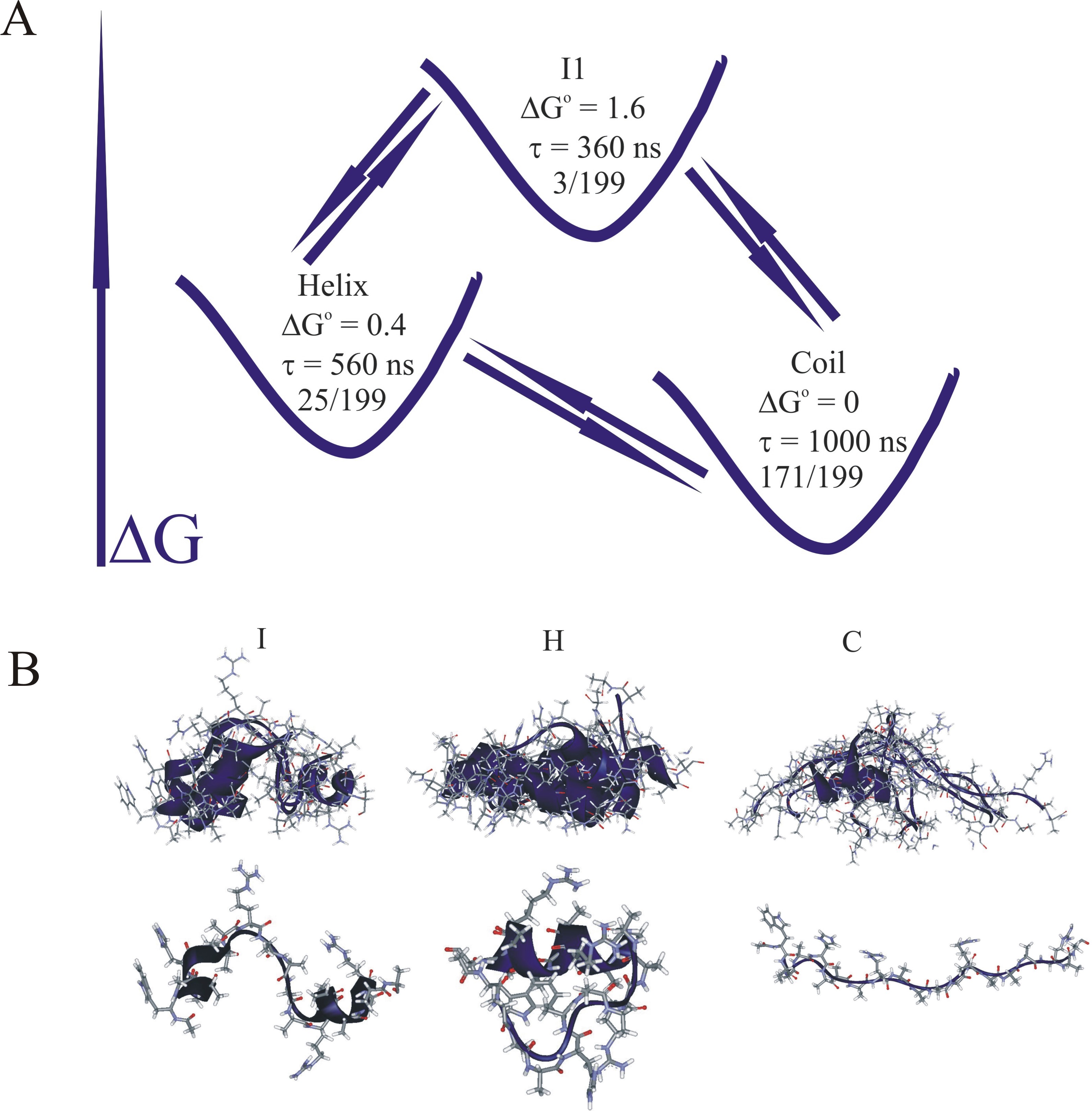
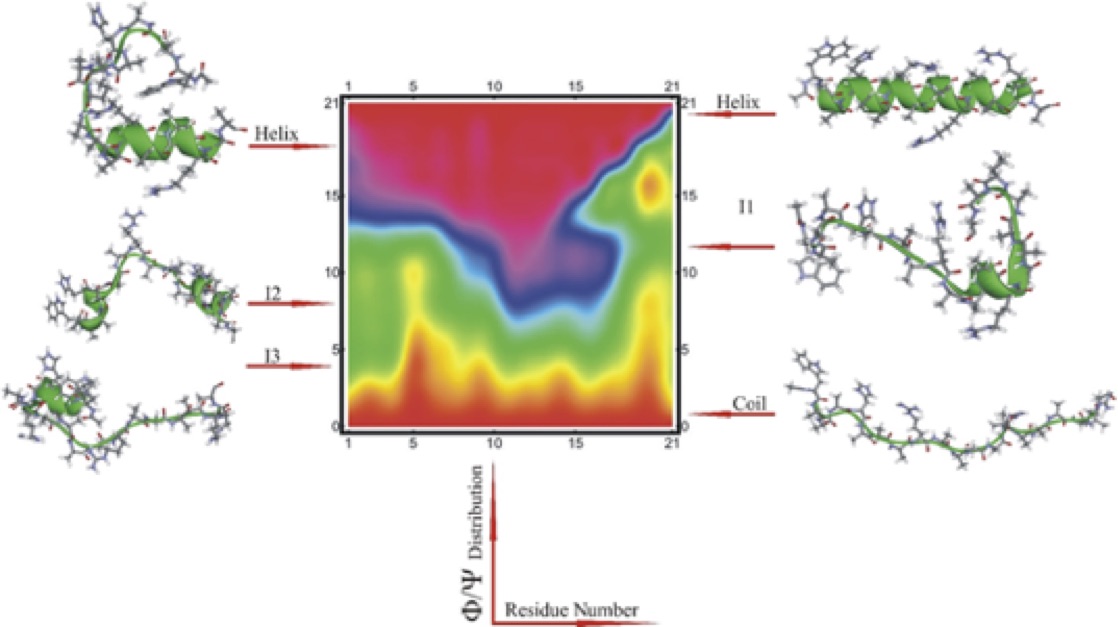
Nanosecond laser temperature jumps with tryptophan fluorescence detection and molecular dynamics simulation with kinetic dimensionality reduction were used to study the helix-coil transition in a 21-residue alpha-helical heteropeptide. Analysis of the temperature dependent relaxation dynamics of this heteropeptide identified a distinct faster component of 20 – 35 ns, besides a slower component of 300-400 ns at temperatures between 296K - 280K. To understand the mechanism of progression from a non-structured coil state to a structured helical state we carried out a 12 microsecond molecular dynamics simulation of this peptide system. Clustering and optimal dimensionality reduction were applied to the molecular dynamics trajectory to generate low-dimensional coarse-grained models of the underlying kinetic network in terms of 2-5 metastable states. In accord with the generally accepted understanding of the multiple conformations and high entropy of the unfolded ensemble of states, the 'coil' metastable set contains the largest number of structures. Interestingly, the helix metastable state was also found to be structurally heterogeneous, consisting of the completely helical form and several partly folded conformers which interconvert at a time scale faster that global folding. The intermediate states contain the fewest structures, have lowest populations and have the shortest lifetimes. As the number of considered metastable states increases, more intermediates and more folding paths appear in the coarse-grained models. One of these intermediates corresponds to the transition state for folding, which involves an 'off-center' helical region over residues 11-16. The kinetic network model is consistent with a statistical picture of folding following a simple reaction coordinate counting the helical population of individual residues. Based on the simulations, we propose that the fast relaxation time should be assigned to cooperative folding/unfolding of segments of 1-4 neighboring residues.
|
|
| WH21 kinetic network with N=3 states. | Statistical folding pathway for WH21. |
_______________________________________________________________________________
G.S. Jas and K. Kuczera
J. Phys. Chem. B in press (2018)
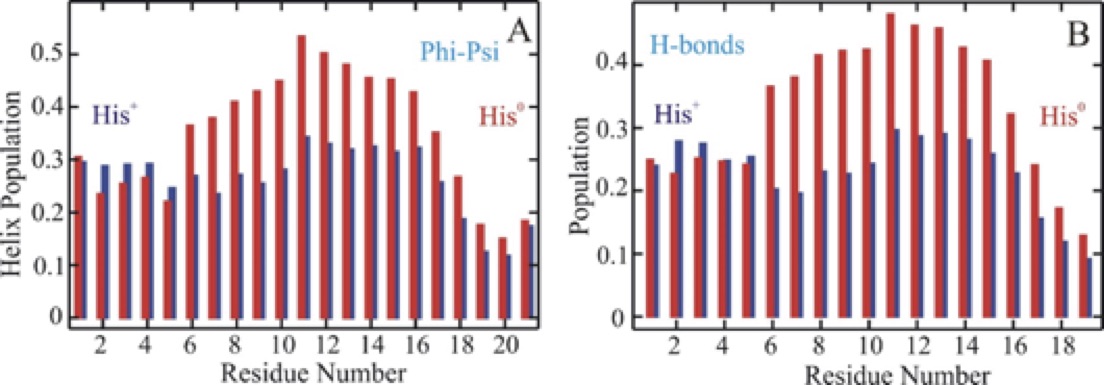
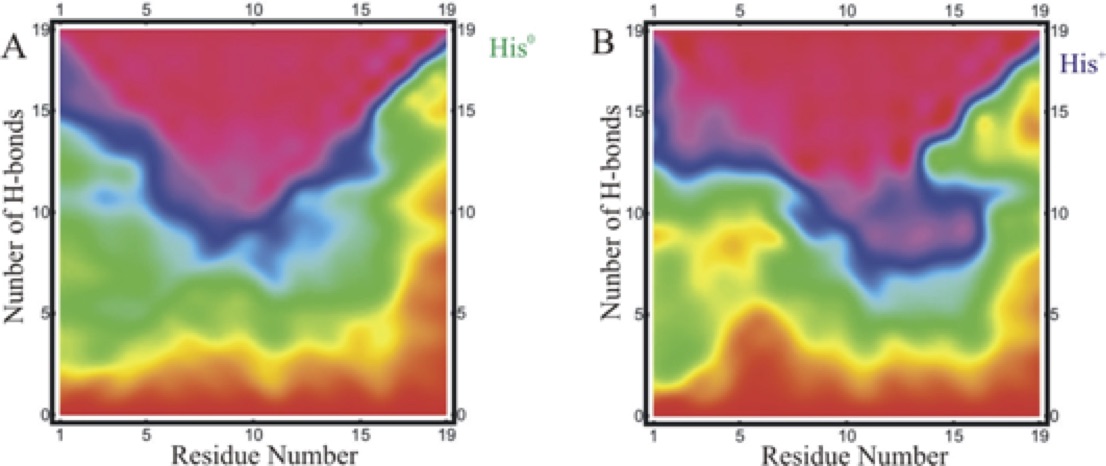
Stability of secondary structural elements is an integral component of a structurally stable protein. Presence of protons in the residue sequence and their immediate environment play a significant role in conformational stability. In this study, we show that removing a proton from a single amino acid residue significantly increases the stability of an alpha helical heteropeptide in comparison with the unprotonated form. Far-UV circular dichroism spectroscopy, fluorescence spectroscopy, fluorescence energy transfer measurements and over ten microseconds of all-atom molecular dynamics simulations are used to provide an atomically detailed characterization of this event. There is a single histidine residue in the studied alpha-helical peptide sequence towards the N-terminal that interacts with a tryptophan located four residues away and quenches the fluorescence when protonated. Removing a proton from this histidine residue dequenches the tryptophan fluorescence and contributes to a significant increase in the helix stability. Atomically detailed analysis of individual residue conformations shows that the protonated histidine tends to be in closer proximity to the tryptophan, which correlates with higher helix content in the N and C termini and lower helix content in the central region of the peptide. In the presence of a neutral histidine, when tryptophan fluorescence is no longer quenched and histidine moves further away from tryptophan, the helix content remains mostly unchanged in the N-and-C termini and significantly increases in the central region. Our results strongly suggest that interactions of the tryptophan with a protonated histidine downregulate the helix population in the central segment of the helical structure compared to a neutral histidine residue. Upregulation of helix population of the central segment of this alpha-helical heteropeptide in the presence of a neutral histidine residue, significantly increases the peptide structural stability.
|
|