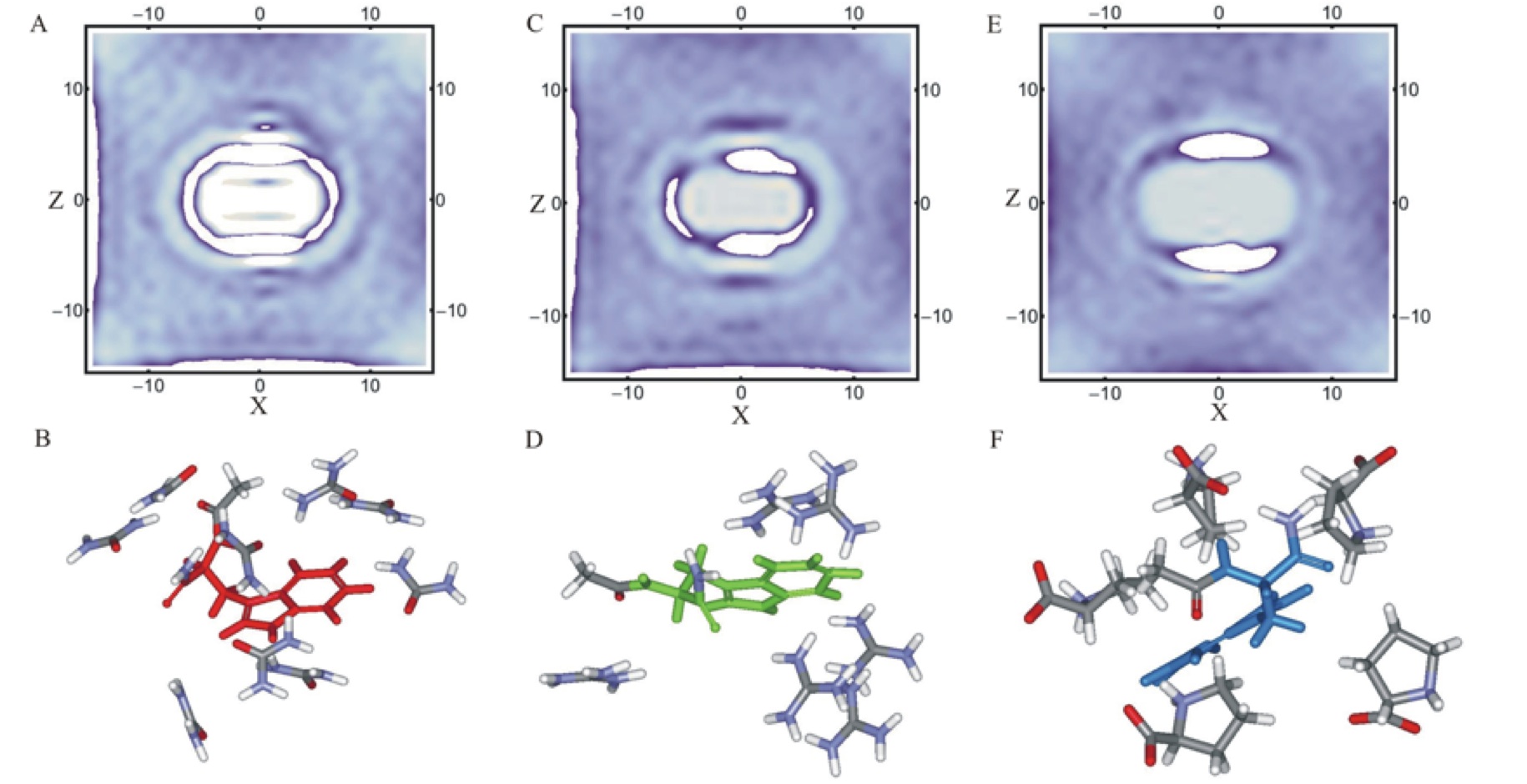
| Three-dimensional co-solvent density and sample solvation patterns of NATA in urea, GdmCl and proline. |
_______________________________________________________________________________
G.S. Jas, C.R. Middaugh and K. Kuczera
J. Phys. Chem. B, 120:6939-6950 (2016)
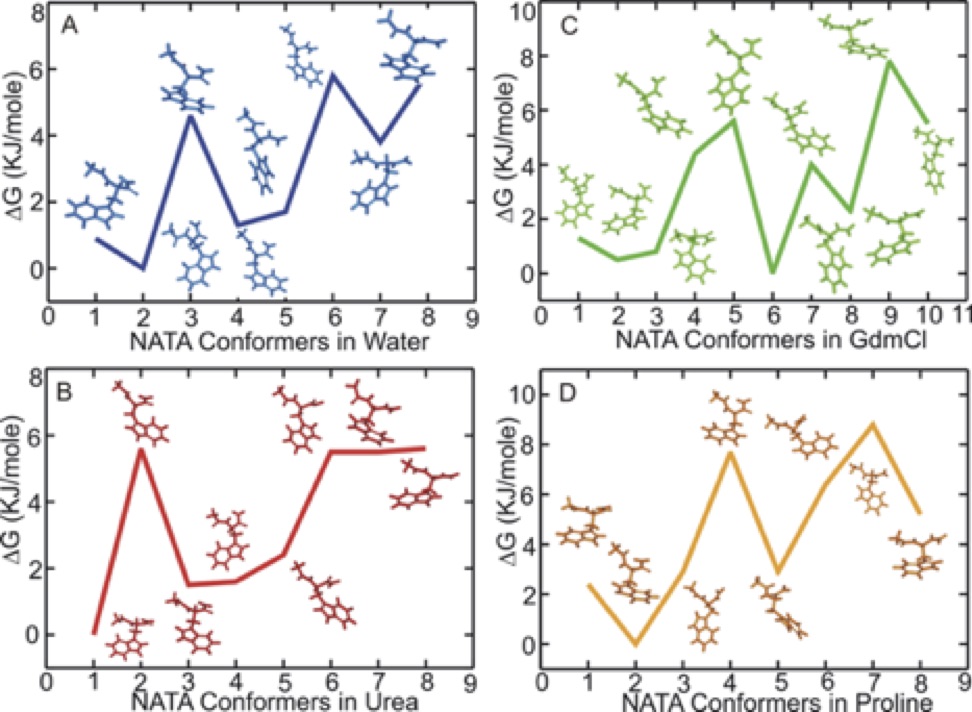
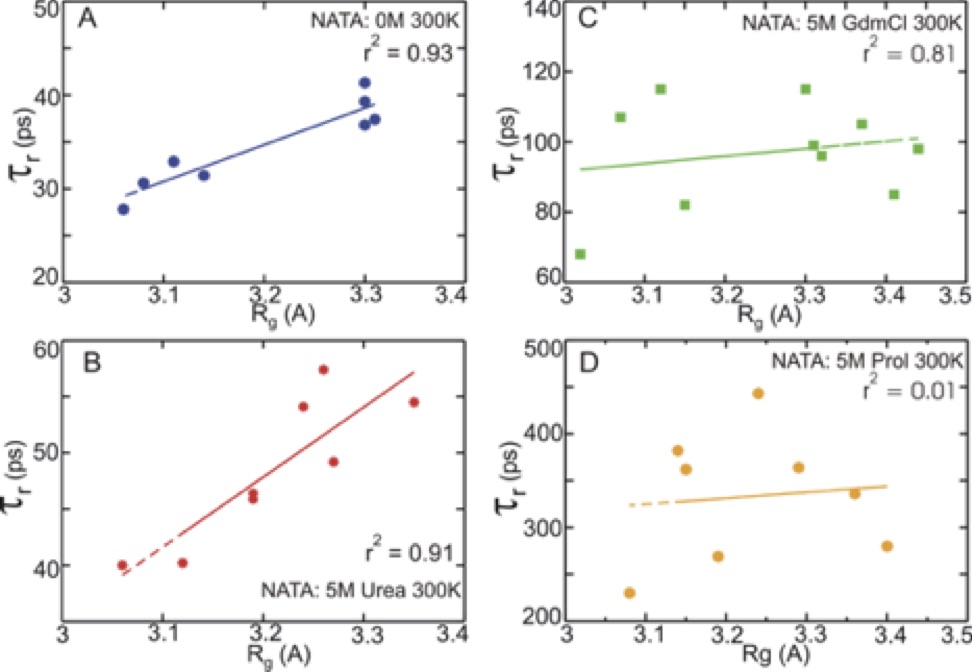
Chaotropes like urea and guanidinium chloride (GdmCl) tend to destabilize, and kosmotropes like proline tend to stabilize folded structures of peptides and proteins. Here, we combine fluorescence anisotropy decay measurements and molecular dynamics simulations to gain a microscopic understanding of the molecular mechanism for shifting conformational preferences in aqueous, GdmCl, urea, and proline solutions of a simple model dipeptide, N-acetyl-tryptophan-amide (NATA). Measured anisotropy decay of NATA as a function of temperature, pH and co-solvent concentrations showed reorientations moderately slower in GdmCl and urea and substantially slower in proline compared to aqueous environment. A small change in pH significantly slows orientation time in water and GdmCl and less markedly in urea. Computationally, we use molecular dynamics with dihedral restraints to separately analyze the motions and interactions of the representative NATA conformers in the four different solvent environments. This novel analysis provides a dissection of the observed overall diffusion rates into contributions from individual dipeptide conformations. The variation of rotational diffusion rates with conformation are quite large. Population-weighted averaging or using properties of the major cluster reproduces the dynamical features of the full unrestrained dynamics. Additionally, we correlate the observable diffusion rates with microscopic features of conformer size, shape and solvation. This analysis uncovered underlying differences in detailed atomistic behavior of the three co-solvents – urea, GdmCl and proline. For both urea and the pure water system we find good agreement with hydrodynamic theory, with diffusion rates primarily correlated with conformer size and shape. In contrast, for GdmCl and proline solutions, the variation in conformer diffusion rates was mostly determined by specific interactions with the co-solvents. We also find preferences for different molecular shapes by the three co-solvents, with increased preferential solvation of smaller and more spherical conformers by urea and larger and more elongated conformers by GdmCl and proline. Additionally, our results provide a basis for a simple approximate model of the effects of pH lowering on dipeptide conformational equilibria. The translational diffusion rates of NATA are less sensitive to conformations, but variation with solvation strength is similar to rotational diffusion. Our results, combining experiment and simulation, show that we can identify the individual peptide conformers with definite microscopic properties of shape, size and solvation, that are responsible for producing physical observables such translational and orientational diffusion in the complex solvent environments of denaturants and osmolytes.
|
|
| NATA structures sampled. | Correlation between rotational time tau and peptide Rg. |
_______________________________________________________________________________
B.L. Lee, K. Kuczera, C.R. Middaugh and G.S. Jas
J. Chem. Phys. 144:245103 (2016)
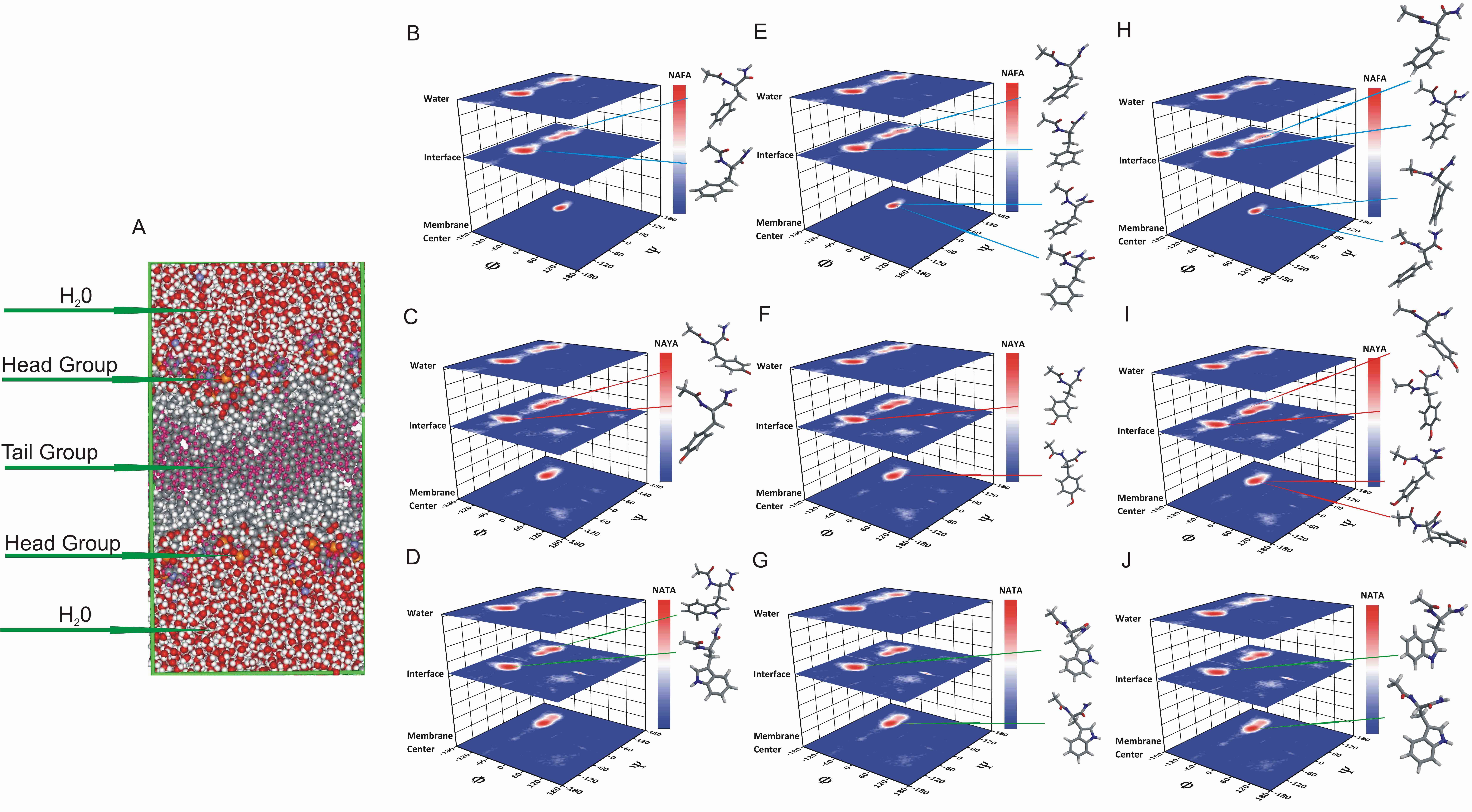
The time-resolved parallel artificial membrane permeability assay with fluorescence detection and comprehensive computer simulations are used to study the passive permeation of three aromatic dipeptides – N-acetyl-phenylalanineamide (NAFA), N-acetyltyrosineamide (NAYA) and N-acetyl-tryptophanamide (NATA) - through a 1,2-dioleoyl-sn-glycero-3-phospocholine (DOPC) lipid bilayer. Measured permeation times and permeability coefficients show fastest translocation for NAFA, slowest for NAYA and intermediate for NATA under physiological temperature and pH. Computationally, we perform umbrella sampling simulations to model the structure, dynamics and interactions of the peptides as a function of z, the distance from lipid bilayer. The calculated profiles of the potential of mean force show two strong effects – preferential binding of each of the three peptides to the lipid interface and large free energy barriers in the membrane center. We use several approaches to calculate the position-dependent translational diffusion coefficients D(z), including one based on numerical solution the Smoluchowski equation. Surprisingly, computed D(z) values change very little with reaction coordinate and are also quite similar for the three peptides studied. In contrast calculated values of sidechain rotational correlation times τrot(z) show extremely large changes with peptide membrane insertion – values become 100 times larger in the headgroup region and 10 times larger at interface and in membrane center, relative to solution. The peptides’ conformational freedom becomes systematically more restricted as they enter the membrane, sampling α and β and C7eq basins in solution, α and C7eq at the interface and C7eq only in the center. Residual waters of solvation remain around the peptides even in the membrane center. Overall, our study provides an improved microscopic understanding of passive peptide permeation through membranes, especially on the sensitivity of rotational diffusion to position relative to the bilayer.
|
| NATA conformations as function of membrane insertion |